因为基因组不同区域的不同组蛋白修饰的生物学意义不一样,所以研究它就不得在不每个项目里面做很多数据,很烧钱,比如 Roadmap 表观计划,通常是:6种 chromatin marks H3K4me3, H3K4me1, H3K36me3, H3K27me3, H3K9me3 and H3K27ac)
我看到一个软件的参数是;-R Genomic *regions* to plot: tss, tes, genebody, exon, cgi, enhancer, dhs or bed ,就是设计好了可以针对基因组的任意功能区域进行探索。
那么,基因组不同区域的不同组蛋白修饰的生物学意义到底该如何理解呢?
建议大家参考2013年npg的一篇review:《Histone modifications for human epigenome analysis》,它介绍了如下所示的不同组蛋白修饰:
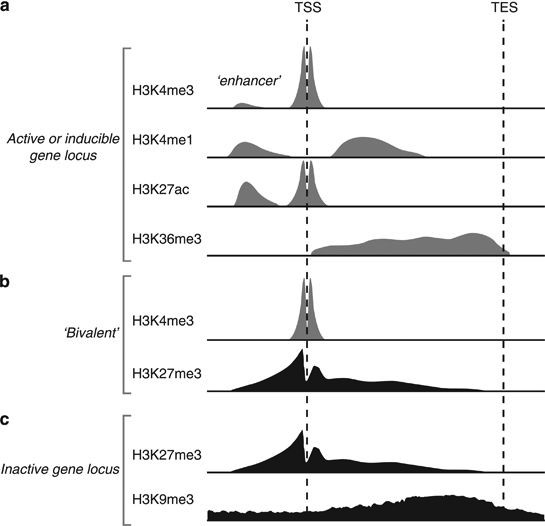
H3K4me3, H3K4me1, H3K36me3, H3K27me3, H3K9me3 and H3K27ac)
可以看到:
- 如果一个基因的启动子区域有H3K27me3修饰,那么它通常是被抑制的
- 如果一个基因的启动子区域有H3K4me3或者H3K27ac修饰,那么它通常是被激活的
- 如果一个基因的body有H3K9me3修饰,那么它通常是被抑制的
- 如果一个基因的body有H3K36me3修饰,那么它通常是被激活的
确实背诵下来有点难哦,大家一起加油哦,选择了生命科学这样的“文科”就不得不熬夜背诵。
通常组蛋白修饰得到的是Chipseq数据,如果要分析: - 首先走上游ChIP-seq流程,即选取唯一比对的bam的序列拿去走MACS2找peaks
- 然后学会deeptools工具探索比对结果,及信号特征:包括信号热图,信号强度profile,样本相关性图
IGV可视化 - 学会 ChIPQC 和 DiffBind 两个R包
- bedtools对bam文件看 全基因组各个染色体的reads密度图
- peaks的数量,长度特特征,基因特征(外显子,内含子,UTR,启动子)注释及motif查找
如果想掌握上面的流程,也可以看我们《生信技能树》的B站免费NGS数据处理视频课程: - 免费视频课程《ChIP-seq数据分析》
- 免费视频课程《ATAC-seq数据分析》