前面的教程:把火山图做成烟花图 ,我们批评了一个学员做差异分析时候的错误的思路,就是把每个分组里面的单个样品简单粗暴的复制粘贴3次充当生物学重复,会导致计算得到的p值如此的诡异。
exprSet=cbind(
exprSet[,1:2],
exprSet[,1:2],
exprSet[,1:2],
exprSet[,1:2]
)
colnames(exprSet)=1:8
那么, 假如你的两个分组真的就是都有且仅有一个样品,你该如何进行差异分析呢?
当然也是有办法的,大家在前面的教程:把火山图做成烟花图 也看到了大量的留言给出了解决方案!我们就摘选其中最简单的edgeR包来分享一下。
首先仍然是从查看表达量矩阵和分组信息
这个时候需要参考前面的教程:把火山图做成烟花图 ,自己制作 Step01-airwayData.Rdata 这个文件,它里面存储了表达量矩阵和分组信息:
rm(list = ls())
options(stringsAsFactors = F)
# 读取基因表达矩阵信息
lname <- load(file = "./data/Step01-airwayData.Rdata")
lname
# 查看分组信息和表达矩阵数据
exprSet <- filter_count
dim(exprSet)
exprSet[1:4,1:4]
group_list
table(group_list)
# 加载包
library(DESeq2)
exprSet = exprSet[,1:2]
group_list=group_list[1:2]
可以看到的是,我们简单粗暴的选取了这个表达量矩阵的前两个样品而已。这样的话,我们的两个分组就各自都有且仅有一个样品啦,这样的无生物学重复的实验设计其实也可以勉强做差异分析。
使用 edgeR包 设置 bcv参数来做无重复的差异分析
代码很简单, 如下所示:
#加载包
library(edgeR)
#设置分组信息
exprSet <- DGEList(counts = exprSet, group = group_list)
bcv = 0.1#设置bcv为0.1
et <- exactTest(exprSet, dispersion=bcv^2)
DEG_edgeR=as.data.frame(topTags(et, n = nrow(exprSet$counts)))
head(DEG_edgeR)
write.csv(DEG_edgeR, 'single-sample-deg-result.csv', quote = FALSE)
前面的教程:把火山图做成烟花图 里面我们展示了 DEseq2和limma两个包,这个时候又出来了 edgeR包 ,其实就大满贯啦。转录组差异分析最常用的3个包,就是它们了。
仍然是绘制火山图
代码如下所示:
#筛选上下调,设定阈值
fc_cutoff <- 1.5
pvalue <- 0.05
DEG_edgeR$regulated <- "normal"
loc_up <- intersect(which(DEG_edgeR$logFC>log2(fc_cutoff)),
which(DEG_edgeR$PValue<pvalue))
loc_down <- intersect(which(DEG_edgeR$logFC < (-log2(fc_cutoff))),
which(DEG_edgeR$PValue<pvalue))
DEG_edgeR$regulated[loc_up] <- "up"
DEG_edgeR$regulated[loc_down] <- "down"
head(DEG_edgeR)
library(AnnoProbe)
ag=annoGene(rownames(DEG_edgeR),
ID_type = 'ENSEMBL',species = 'human'
)
head(ag)
DEG_edgeR$ENSEMBL=rownames(DEG_edgeR)
deg_anno=merge(ag,DEG_edgeR,by='ENSEMBL')
head(deg_anno)
library(EnhancedVolcano)
colnames(deg_anno)
EnhancedVolcano(deg_anno,
lab =deg_anno$SYMBOL,
x = 'logFC',
y = 'FDR')
出图如下所示:

可以看到这个时候的火山图其实是非常正常的!
可以跟原始的差异分析做对比
首先查看两次差异分析:
reulsts_edgeR = deg_anno
colnames(reulsts_edgeR)
load( file = "./data/Step03-DESeq2_nrDEG.Rdata")
reulsts_DEseq2 = deg_anno
colnames(reulsts_DEseq2)
df=merge(reulsts_edgeR[,c(1,2,7,10,11)],
reulsts_DEseq2[,c(1,8,12,13)],
by='ENSEMBL')
table(df$regulated.x,df$regulated.y)
如下所示:
> colnames(reulsts_edgeR)
[1] "ENSEMBL" "SYMBOL" "biotypes" "chr" "start" "end"
[7] "logFC" "logCPM" "PValue" "FDR" "regulated"
> colnames(reulsts_DEseq2)
[1] "ENSEMBL" "SYMBOL" "biotypes" "chr"
[5] "start" "end" "baseMean" "log2FoldChange"
[9] "lfcSE" "stat" "pvalue" "padj"
[13] "regulated"
需要把两个差异分析结果矩阵合并,然后对比!
down normal up
down 0 355 670
normal 424 13643 860
up 719 789 0
可以看到, 上下调居然是反过来了,哈哈哈,可能是前面的代码没有设置因子。
我们可以散点图更加直观查看,代码如下所示:
library(ggplot2)
library(ggpubr)
library(ggstatsplot)
p <- ggplot(df, aes(x=df$logFC,
df$log2FoldChange))+
geom_point()+
labs(x = "logFC",
y = "log2FoldChange")+
scale_color_manual(values=c("blue", "grey","red"))+
geom_vline(xintercept = c( -1.5,0,1.5),lty=4,col="#ec183b",lwd=0.8) +
geom_hline(yintercept = c(-1.5,0,1.5 ),lty=4,col="#18a5ec",lwd=0.8) +
#xlim(-5,5)+
#ylim(-3,3)+
theme_ggstatsplot()+
theme(panel.grid.minor = element_blank(),
panel.grid.major = element_blank())
p
效果如下:
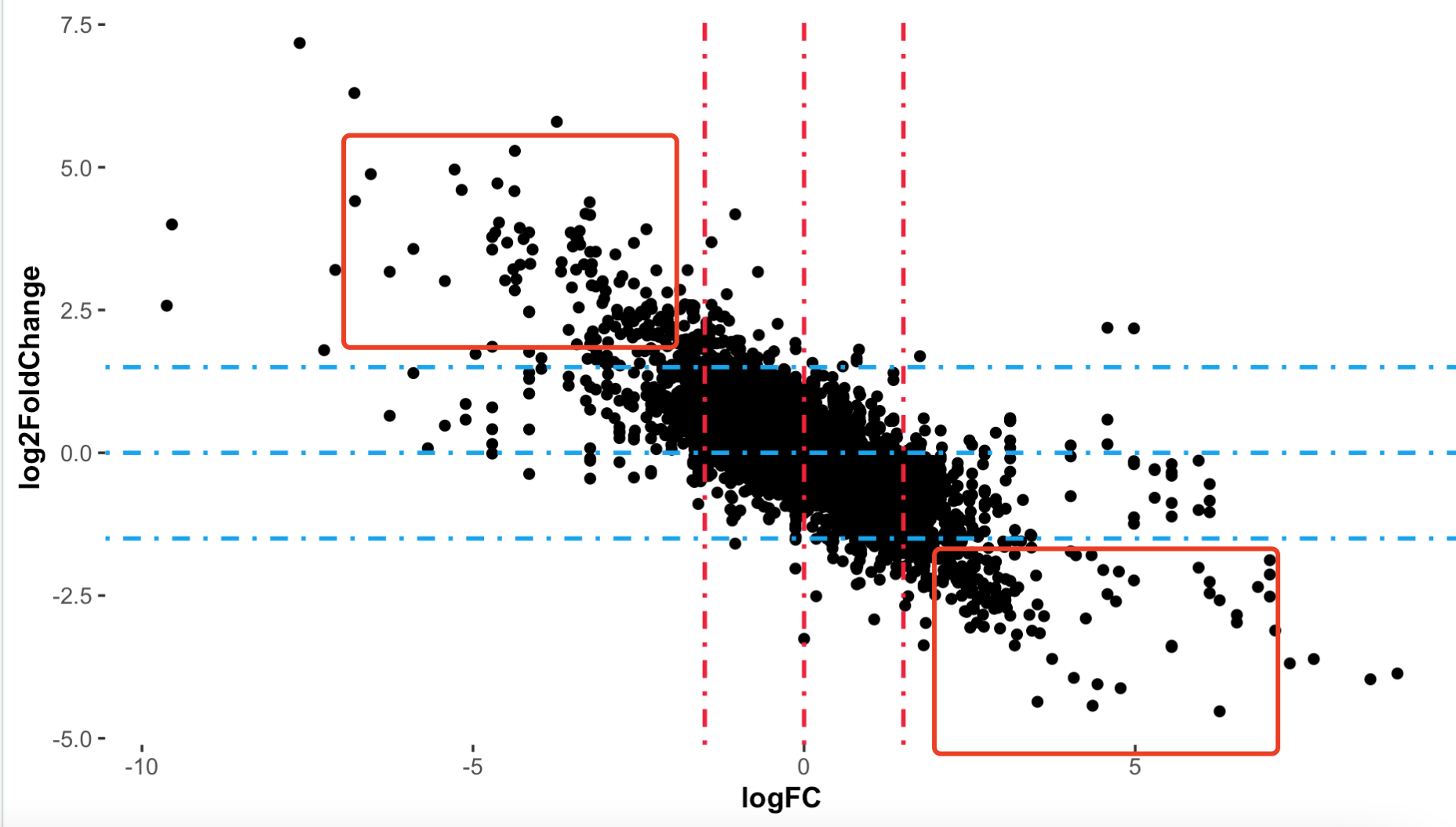
还有个韦恩图,也可以查看两次差异分析结果的区别:
代码如下所示:
library(data.table)
library(ggplot2)
library(ggstatsplot)
library(ggvenn)
a <- list( 'edgeR_up'=split(df$ENSEMBL,df$regulated.x)$up,
'edgeR_down'=split(df$ENSEMBL,df$regulated.x)$down,
'DEseq2_up'=split(df$ENSEMBL,df$regulated.y)$up,
'DEseq2_down'=split(df$ENSEMBL,df$regulated.y)$down
)
p1 <- ggvenn(a,
fill_alpha = 0.9,
fill_color = c('#7fc97f','#beaed4','#fdc086','#ffff99'),
set_name_size = 5,
stroke_size = 0.5)
p1
出图如下:
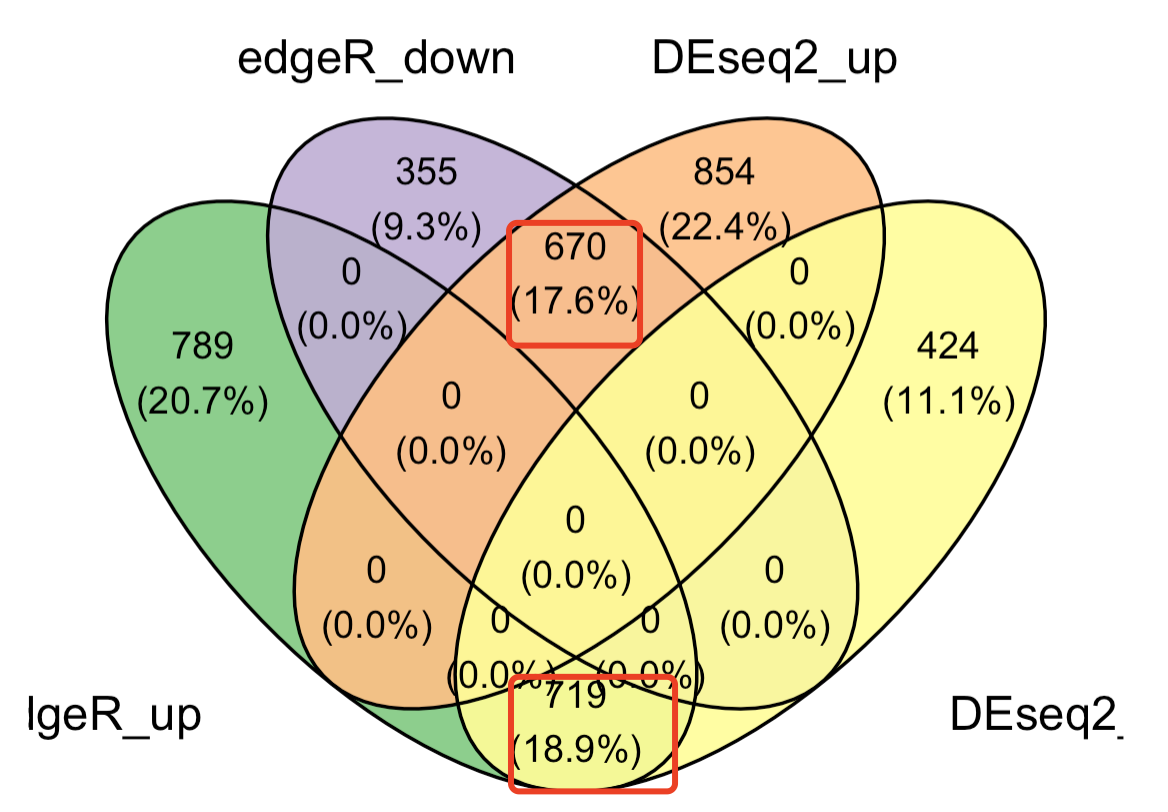
因为我们前面两次 差异分析的因子没有设置,所以这个上下调是反过来的,但是仍然是可以看到,两次差异分析的结果的一致性比较好!
学徒作业
运行我的代码,把因子设置好,重新进行散点图,韦恩图的绘制!