以 Clinical & Translational Immunology 2021; 文章为例,标题是:《Patient-derived organoids of bladder cancer recapitulate antigen expression profiles and serve as a personal evaluation model for CAR-T cells in vitro》,是中国深圳的中科院先进院的团队的研究成果。
跟普通的肿瘤外显子测序数据的分析步骤并没有较大差异,如下所示:

虽然这个研究仅仅是产生了3个类器官数据,但是需要有9个样品进行外显子测序哦,其中每个病人的正常对照仅仅是数据分析过程使用,并不需要在后续可视化进行呈现。对类器官数据的可视化主要是比较样品培养前的肿瘤组织和培养后的类器官组织的突变结果的一致性, 首先看SNV;
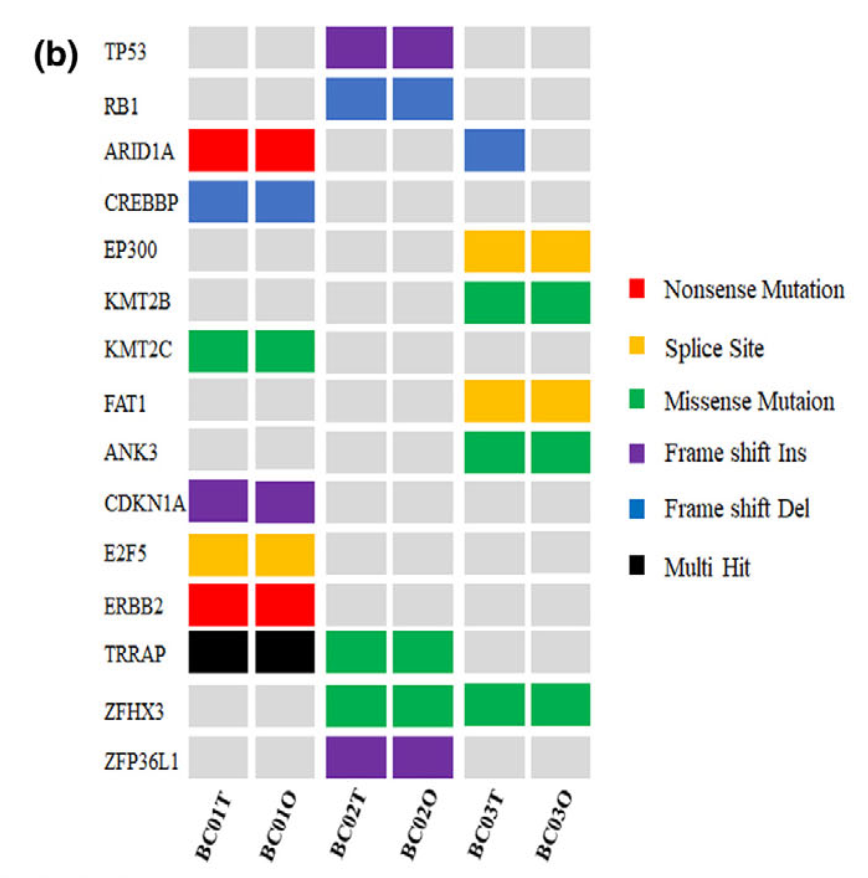
可以看到, 绝大部分的SNV在每个样品培养前的肿瘤组织和培养后的类器官组织的突变结果具备非常好的一致性。包括上图显示的各种突变形式:
- onsense mutations (e.g. ARID1A and ERBB2)
- missense mutations
- splice site mutations (e.g. EP300 and FAT1)
- multiple hit mutations
- frame shift indels/dels (e.g. TP53, RB1,CREBBP and CDKN1A)
其实就是maftools的标准图表啦:
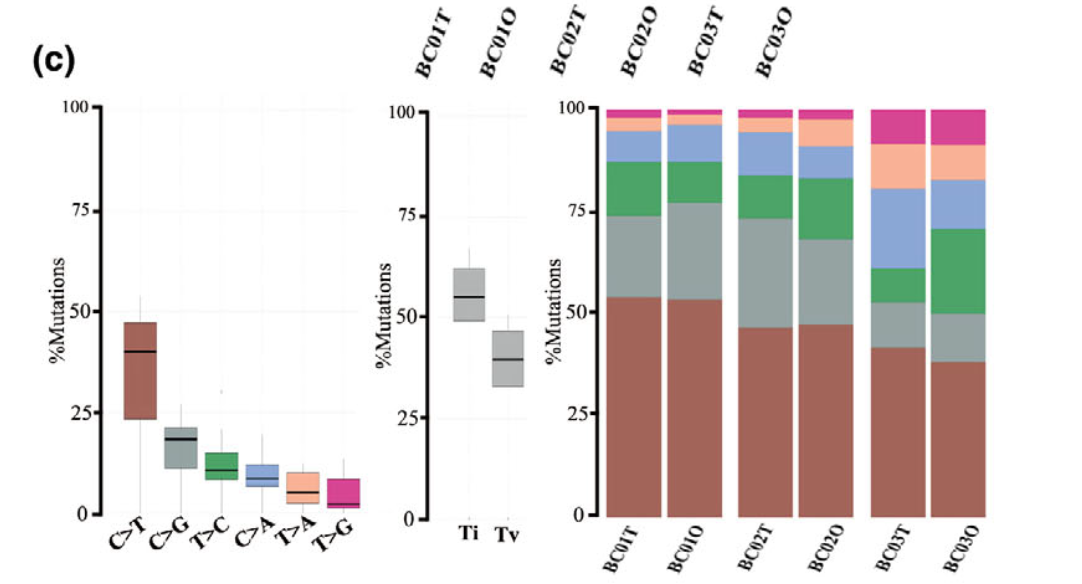
还有CNV的对比:

样本量多少都是类似的分析
总之就是要说明自己的 organoids recapitulate the genetic alterations in the parental tumors. 既然是外显子测序,那就是从肿瘤外显子的SNV和CNV两个分析结果的角度来说明。
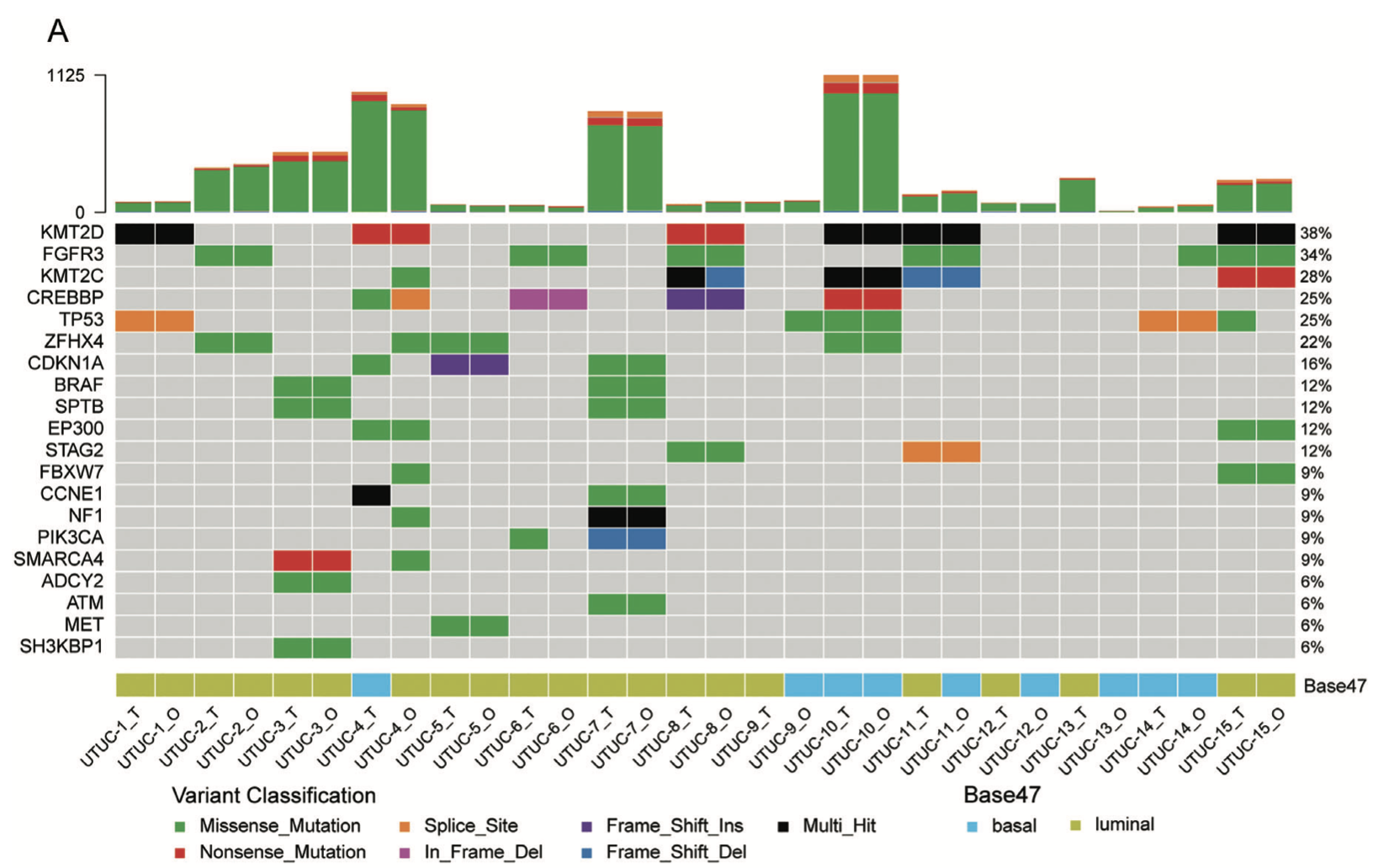
再比如:2021年12月17日,北京大学第一医院泌尿外科联合深圳市第二人民医院在Advanced Science期刊 (IF=16.8) 在线发表了标题为《Patient-derived Upper Tract Urothelial Carcinoma Organoids as a Platform for Drug Screening》的研究成果,也是这样的展现:
首先看:A) Somatic genomic landscape of 15 UTUC organoid lines (_O) and the corresponding parental tumors (_T). 仍然是 maftools的标准图表 :
另外,他们在展现CNV的一致性,应该是采用了发表在Gigascience . 2021 May的工具《MesKit: a tool kit for dissecting cancer evolution of multi-region tumor biopsies through somatic alterations》:
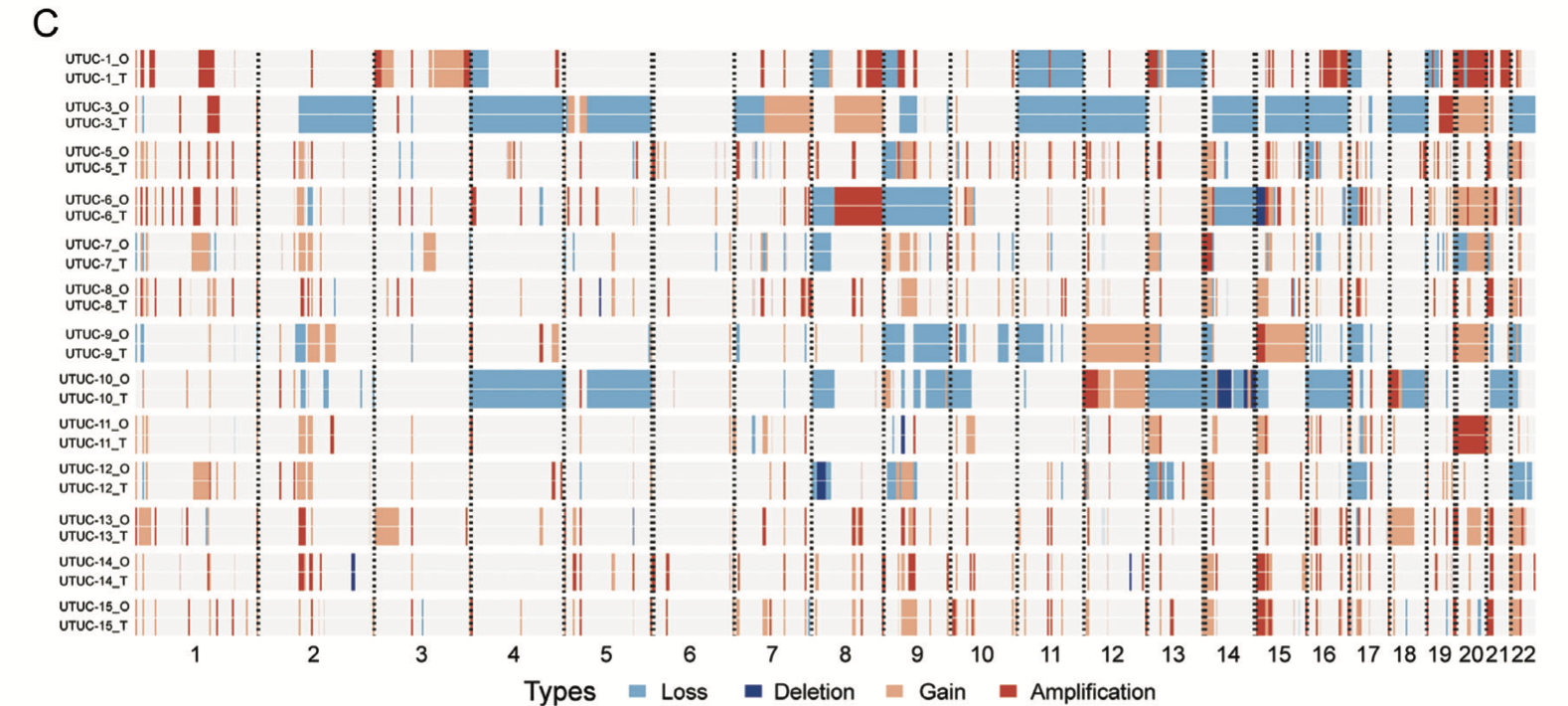
值得玩味的是文章里面并没有提到 这个MesKit工具,我在原文里面看了看:https://onlinelibrary.wiley.com/doi/full/10.1002/advs.202103999 ,其提到的肿瘤外显子数据分析流程里面的软件工具列表如下所示:
- Fastp (v0.12.6) was used to remove adaptors and filter low quality reads.[39]
- Single-nucleotide variant (SNVs) were called by GATK(v4.1.9).[40]
- Sequence reads were mapped against the human reference genome (hg38) using Burrows-Wheeler Alignment with maximal exact matches (BWA-MEM) v0.7.12.[41]
- Read mapping was followed by the marking of duplicates, merging of lanes, and realignment of indels using Picard (v2.23.8).
- Somatic SNVs and indels in the tumors and organoids were called with Mutect2 (default options) using the matched blood samples as the reference.
- Somatic SNVs with a VAF < 0.05 and supported by less than 3 reads were filtered out.
- Mutation effect predictions and annotations were performed by VEP (release 101).[42]
- CNVs were detected using TitanCNA (1.30.0).[43]
- The clone architecture and the VAF of mutated sites in organoids and the corresponding tumors were inferred by sciClone.[44]
不过,也有可能作者确实并没有使用 这个MesKit工具,而是自己写代码进行了前面的CNV一致性的可视化展现呢!
如果你也对这方面数据分析感兴趣,我们《生信菜鸟团》公众号有一个《肿瘤基因组测序数据分析专栏》,精选部分目录如下:
- CNVkit的使用方法
- Strelka2 call Somatic 流程
- GATK 的 Germline mutation 流程
- 肿瘤外显子测序数据分析公开课
- Hallmarks | 癌症的特征II
- GATK 的Somatic Mutation流程
- 肿瘤基因组测序数据分析专栏—前言
- 肿瘤基因组测序数据高级分析
- 文献阅读 · 变异分析流程
- 标志| 癌症的特征
- 基础知识 · ClinVar
- 基础知识 · COSMIC 数据库
- 基础知识 · ExAC、GnomAD、1000G、dbSNP
- 基础知识 · 常见数据库
- 基础知识 · 全基因组测序WGS
- 基础知识 · 全外显子测序WES
- 基础知识 · 靶向测序Panel