公众号后台看到了很多人提问如何安装一个免疫治疗数据队列包, IMvigor210CoreBiologies ,因为它是一个100多M的压缩包文件,在 http://research-pub.gene.com/IMvigor210CoreBiologies/packageVersions/ 网页可以看到:
IMvigor210CoreBiologies_0.1.13.tar.gz 15-Feb-2018 10:56 116M
IMvigor210CoreBiologies_1.0.0.tar.gz 11-Feb-2019 20:49 116M
实际上,但凡学过一点点R语言的,都知道如何下载这样的R语言源代码压缩包文件来进行安装。实际上,这个包的 的官方说明书也写的很清楚:http://research-pub.gene.com/IMvigor210CoreBiologies/
因为这个包是旧版本的R和bioconductor时代创造的
所以它官方教程写的是下面的安装方法:
First load the utility:
source("http://bioconductor.org/biocLite.R")
Now copy the following into your R session to load mandatory dependencies:
pkg = c("biomaRt",
"circlize",
"ComplexHeatmap",
"corrplot",
"DESeq2",
"dplyr",
"DT",
"edgeR",
"ggplot2",
"limma",
"lsmeans",
"reshape2",
"spatstat",
"survival",
"plyr")
biocLite(pkg)
上面的代码是过时的哦!应该是我们周末班准备工作给的安装R包 http://www.bio-info-trainee.com/3727.html
最新的批量安装R包的方式
首先配置中国大陆特色镜像
options()$repos
options()$BioC_mirror
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
options()$repos
options()$BioC_mirror
然后使用下面的代码,进行批量安装:
list.of.packages <- c("shiny",
"tidyr",
'tidyverse',
"clusterProfiler",
"DT",
"ashr",
"enrichplot",
"plotly")
all_packages = rownames(installed.packages())
save(all_packages,file = 'all_packages.Rdata')
#checking missing packages from list
new.packages <- list.of.packages[!(list.of.packages %in% installed.packages()[,"Package"])]
new.packages
packToInst <- setdiff(list.of.packages, installed.packages())
packToInst
if(T){
lapply(packToInst, function(x){
BiocManager::install(x,ask = F,update = F)
})
}
lapply(intersect(packagesReq, installed.packages()),function(x){
suppressPackageStartupMessages(library(x,character.only = T))
})
其实你有没有发现,代码反而是多了呢?
接下来看 IMvigor210CoreBiologies_1.0.0.tar.gz这个116M的文件
必须要有前面的依赖包,然后才能下载 IMvigor210CoreBiologies_1.0.0.tar.gz这个116M的文件,进行安装,代码如下所示:
Finally, install IMvigor210CoreBiologies itself. Assuming that you’re already running R in the directory containing the downloaded package file,
install.packages("IMvigor210CoreBiologies_1.0.0.tar.gz",
repos=NULL)
As usual, once the package is installed on your system, you also need to load it into your active R session:
library("IMvigor210CoreBiologies")
但是你仍然是发现缺失 DESeq这个包吗
而我们的 IMvigor210CoreBiologies 依赖于它DESeq这个包,所以仍然是失败的,只能是IMvigor210CoreBiologies这个包写的有点问题, 应该是它本来就并不是为了发布它,所以也无所谓,这样的粗制滥造的包本来就是不可能过审,没办法发布的。
实际上,对于这样的包,原则上我们首先应该是摒弃它。如果你一定要安装使用它,我们有两个解决方案,首先,你打开 IMvigor210CoreBiologies_1.0.0.tar.gz这个116M的文件 ,进去修改这个包的依赖情况。就是这个改 DESCRIPTION 文件里面的R版本依赖 ,可以看到里面有 DESeq, DESeq2,等包,其实没有必要依赖这些啊, 删除 DESeq即可,修改好文件后,重新安装这个包:
Depends: R (>= 3.3), Biobase
Imports: biomaRt, circlize, ComplexHeatmap, corrplot, DESeq, DESeq2,
dplyr, DT, edgeR, ggplot2, graphics, limma, lsmeans, methods,
reshape2, spatstat, stats, survival, plyr
Suggests: roxygen2
License: file LICENSE
超级简单!这样你可以很顺利的安装它,但是因为你强行修改了它的依赖,所以你安装的应该是一个残缺版本,如果后续它这个包确实是依赖于 DESeq 包里面的函数或者对象,就尴尬了。
另外一个选择是,你压根就没有必要去安装这个包,因为你仅仅是需要它里面的数据,你再仔细看教程,其实就是:To load a CountDataSet object called ‘cds’, type:
data(cds)
This CountDataSet object contains raw counts for all genes as well as basic feature and all sample annotations reported in the manuscript. These can be accessed like this:
head(counts(cds))
head(fData(cds))
head(pData(cds))
如果你是熟悉R包结构,就明白它自带的数据,其实就存储在 data 文件夹:
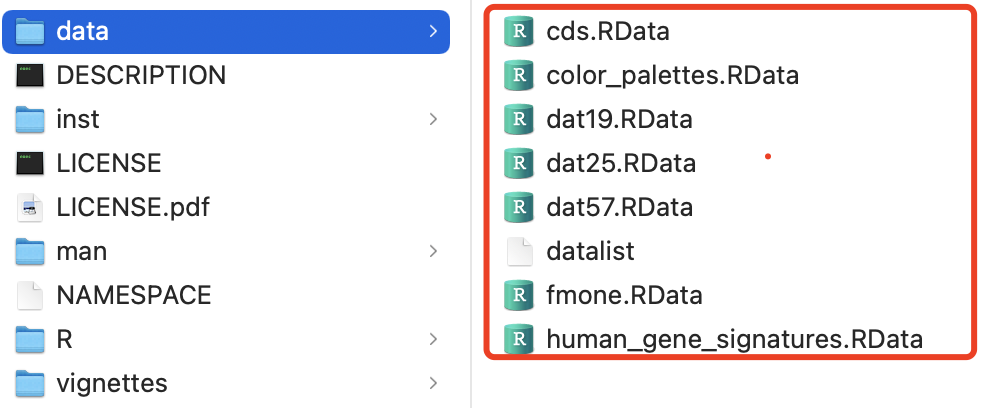
你不需要安装这样的包,也不需要加载它,仅仅是load这个压缩包里面的文件夹里面的对应的R数据对象文件即可。
但是,尴尬的是它这个对应的R数据对象文件里面的 cds变量其实是一个 CountDataSet object ,这个对象来自于 DESeq 包,如果你没办法按照它这个DESeq包,就不可能使用它定义的CountDataSet对象,那么你就无法load对应的R数据对象文件成功,报错如下:
> cds
载入需要的程辑包:DESeq
Error in .requirePackage(package) :
unable to find required package ‘DESeq’
此外: Warning message:
In library(package, lib.loc = lib.loc, character.only = TRUE, logical.return = TRUE, :
不存在叫‘DESeq’这个名字的程辑包
载入需要的程辑包:DESeq
收捲时出错: unable to find required package ‘DESeq’
Error: no more error handlers available (recursive errors?); invoking 'abort' restart
这个问题就麻烦了,夜已深了,先暂停吧!