RNA-seq数据分析我们分享了很多,有RNA-seq数据分析经验的小伙伴都会觉得很简单,直接把fastq格式的测序数据比对到合适的参考基因组,然后根据匹配的基因组注释文件去定量就可以拿到表达量矩阵。后续就是基于表达量矩阵的差异分析,富集分析!
但是如果RNA-seq数据分析项目非常多,或者说每个项目里面的样品非常多, 这个时候我们会推荐流程化管理我们的脚本,我个人的数据分析生涯主要是shell脚本,因为并不是企业级项目管理,能跑十几个项目还是因为要去给粉丝帮忙。对企业生产实践来说,Snakemake流程化管理各个NGS数据分析流程是一个很好的选择,恰好看到了一个最新的 Snakemake workflow, 推荐给大家。
就是:ARMOR (Automated Reproducible MOdular RNA-seq) is a Snakemake workflow,
这个流程图里面的软件我都很喜欢,在我们公众号的1.3万篇教程里面,可以搜索到这个流程图里面的涉及到的每个软件的具体讲解 :
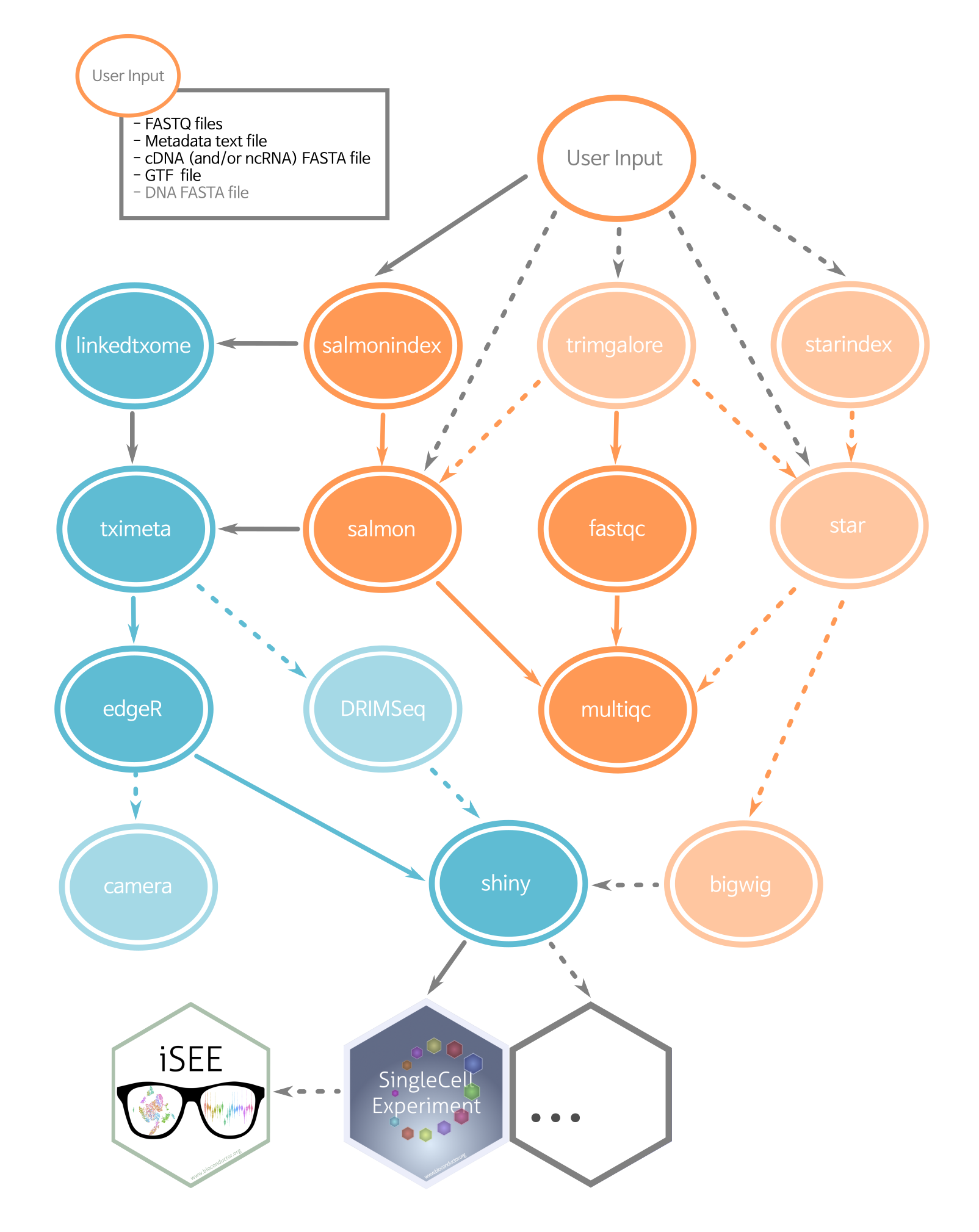
对这个流程代码感兴趣的可以自己去看 ARMOR (Automated Reproducible MOdular RNA-seq) 的GitHub主页哦!
我这里就不赘述啦!
另外,附上我自己的RNA-seq数据分析shell脚本实践
如果是真实的转录组项目,每个步骤耗费计算机资源和时间都很可观,这个时候可以采用模拟数据来测试流程和代码。下面是我的示例代码,如果看懂,需要有一定的Linux基础知识哦:
首先对项目测序数据取2500条reads
代码如下:
mkdir -p $HOME/rna/test/small_raw/
# 在 23.GSE149638 这个公共数据集里面是96个双端测序的转录组数据
cd $HOME/project/23.GSE149638/raw
# 取2500条reads
ls *gz|while read id ;do ( zcat $id |head -10000 | gzip -c - > $HOME/rna/test/small_raw/$id );done
cd $HOME/rna/test/small_raw/
raw 测序数据
为了更进一步加快速度,可以直接测试6个样品即可。
ls -lh |cut -d" " -f 5-
197K 2月 20 10:36 SRR10574381_1.fastq.gz
208K 2月 20 10:36 SRR10574381_2.fastq.gz
198K 2月 20 10:36 SRR10574382_1.fastq.gz
201K 2月 20 10:36 SRR10574382_2.fastq.gz
206K 2月 20 10:36 SRR10574383_1.fastq.gz
-------
trim
因为是双端测序,所以trim_galore命令需要加上—paired 的参数哦!
如果样本量比较少,就直接 nohup到后台 :
cd $HOME/rna/test/small_raw
mkdir ../cleanData
ls *gz|cut -d"_" -f 1|sort -u |while read id;do
nohup trim_galore -q 25 --phred33 --length 36 --stringency 3 --paired -o ../cleanData ${id}*.gz &
done
如果样本量比较多,就控制一下每次并行的时候项目提交的数量:
ls *gz|cut -d"_" -f 1|sort -u |while read id;do
echo trim_galore -q 25 --phred33 --length 36 --stringency 3 --paired -o ../cleanData ${id}_*.gz
done > trim_galore.sh
for i in {0..29};do ( nohup bash ../submit.sh trim_galore.sh 30 $i 1>log.trim_galore.$i.txt 2>&1 & ) ;done
# 如果是单端 :
ls *gz| sort -u |while read id;do
echo trim_galore -q 25 --phred33 --length 36 --stringency 3 -o ../cleanData ${id}
done > trim_galore.sh
这样就是每次并行10个样品,相当于批处理啦,其中 submit.sh 脚本内容如下所示;
cat $1 |while read id
do
if((i%$2==$3))
then
$id
fi # check the number1 number2
i=$((i+1))
done
# for i in {0..9};do ( nohup bash ../submit.sh trim_galore.sh 10 $i 1>log.trim_galore.$i.txt 2>&1 &) ;done
全部的 trim_galore 命令运行完毕后,得到的clean的fq文件如下:
$ ls -lh *gz |cut -d" " -f 5-
173K 2月 20 10:40 SRR10574381_1_val_1.fq.gz
188K 2月 20 10:40 SRR10574381_2_val_2.fq.gz
177K 2月 20 10:40 SRR10574382_1_val_1.fq.gz
183K 2月 20 10:40 SRR10574382_2_val_2.fq.gz
152K 2月 20 10:40 SRR10574383_1_val_1.fq.gz
160K 2月 20 10:40 SRR10574383_2_val_2.fq.gz
71K 2月 20 10:40 SRR10574384_1_val_1.fq.gz
----
这个时候,还可以使用 fastqc软件看看raw和clean的fastq软件的测序质量情况。
hisat 比对
cd ~/rna/test/cleanData
indexPrefix=/home/data/server/reference/index/hisat/hg38/genome
ls *gz|cut -d"_" -f 1|sort -u |while read id;do
nohup hisat2 -p 1 -x $indexPrefix -1 ${id}*_1_val_1.fq.gz -2 ${id}*_2_val_2.fq.gz -S ${id}.hisat.sam &
done
# sam文件转bam
ls *.sam|while read id ;do (nohup samtools sort -O bam -@ 2 -o $(basename ${id} ".sam").bam ${id} & );done
# 这个过程会输出大量中间文件
rm *.sam
# 为bam文件建立索引
ls *.bam |xargs -i samtools index {}
同理,如果是样本数量太多了,就需要考虑并行并且控制提交样品数量
indexPrefix=/home/data/server/reference/index/hisat/hg38/genome
ls -lh $indexPrefix*
ls *gz|cut -d"_" -f 1|sort -u |while read id;do
if((i%$2==$3))
then
hisat2 -p 2 -x $indexPrefix -1 ${id}*_1_val_1.fq.gz -2 ${id}*_2_val_2.fq.gz -S ${id}.hisat.sam
samtools sort -O bam -@ 2 -o ${id}.hisat.bam ${id}.hisat.sam
rm ${id}.hisat.sam
samtools index ${id}.hisat.bam
fi # check the number1 number2
i=$((i+1))
done
# for i in {0..9};do ( nohup bash run_hisat2.sh t 10 $i 1>log.hisat2.$i.txt 2>&1 & ) ;done
得到的sam文件如下:
$ ls -lh *sam |cut -d" " -f 5-
1.8M 2月 20 11:01 SRR10574381.hisat.sam
1.8M 2月 20 11:01 SRR10574382.hisat.sam
1.5M 2月 20 11:01 SRR10574383.hisat.sam
677K 2月 20 11:01 SRR10574384.hisat.sam
1.8M 2月 20 11:02 SRR10574385.hisat.sam
149K 2月 20 11:01 SRR10574386.hisat.sam
1.9M 2月 20 11:01 SRR10574387.hisat.sam
1.8M 2月 20 11:01 SRR10574388.hisat.sam
1.9M 2月 20 11:01 SRR10574389.hisat.sam
1.7M 2月 20 11:01 SRR10574390.hisat.sam
1.8M 2月 20 11:01 SRR10574391.hisat.sam
1.6M 2月 20 11:01 SRR10574392.hisat.sam
1.8M 2月 20 11:01 SRR10574393.hisat.sam
1.8M 2月 20 11:01 SRR10574394.hisat.sam
1.8M 2月 20 11:01 SRR10574395.hisat.sam
1.8M 2月 20 11:01 SRR10574396.hisat.sam
1.9M 2月 20 11:01 SRR10574397.hisat.sam
1.8M 2月 20 11:01 SRR10574398.hisat.sam
1.8M 2月 20 11:01 SRR10574399.hisat.sam
1.8M 2月 20 11:01 SRR10574400.hisat.sam
1.9M 2月 20 11:01 SRR10574401.hisat.sam
1.9M 2月 20 11:01 SRR10574402.hisat.sam
1.8M 2月 20 11:01 SRR10574403.hisat.sam
1.8M 2月 20 11:01 SRR10574404.hisat.sam
得到的bam文件如下:
$ ls -lh *bam |cut -d" " -f 5-
483K 2月 20 11:27 SRR10574381.hisat.bam
483K 2月 20 11:27 SRR10574382.hisat.bam
407K 2月 20 11:27 SRR10574383.hisat.bam
195K 2月 20 11:27 SRR10574384.hisat.bam
432K 2月 20 11:27 SRR10574385.hisat.bam
42K 2月 20 11:27 SRR10574386.hisat.bam
439K 2月 20 11:27 SRR10574387.hisat.bam
460K 2月 20 11:27 SRR10574388.hisat.bam
504K 2月 20 11:27 SRR10574389.hisat.bam
486K 2月 20 11:27 SRR10574390.hisat.bam
485K 2月 20 11:27 SRR10574391.hisat.bam
391K 2月 20 11:27 SRR10574392.hisat.bam
481K 2月 20 11:27 SRR10574393.hisat.bam
466K 2月 20 11:27 SRR10574394.hisat.bam
462K 2月 20 11:27 SRR10574395.hisat.bam
475K 2月 20 11:27 SRR10574396.hisat.bam
497K 2月 20 11:27 SRR10574397.hisat.bam
430K 2月 20 11:27 SRR10574398.hisat.bam
481K 2月 20 11:27 SRR10574399.hisat.bam
424K 2月 20 11:27 SRR10574400.hisat.bam
492K 2月 20 11:27 SRR10574401.hisat.bam
493K 2月 20 11:27 SRR10574402.hisat.bam
485K 2月 20 11:27 SRR10574403.hisat.bam
488K 2月 20 11:27 SRR10574404.hisat.bam
走featureCounts定量流程
featureCounts命令的参数多种多样,可以设置基于基因的ID或者名字来进行计算表达量,或者基于外显子来。
gtf=$HOME/rna/SUPPA2/gtf/gencode.v37.annotation.gtf
# gtf=/home/yb77613/reference/gtf/gencode/gencode.vM12.annotation.gtf
ls -lh $gtf
conda activate rna
nohup featureCounts -T 5 -p -t exon -g gene_id -a $gtf \
-o all.id.txt *.bam 1>counts.id.log 2>&1 &
nohup featureCounts -T 5 -p -t exon -g gene_name -a $gtf \
-o all.name.txt ../align/*bam 1>counts.name.log 2>&1 &
仔细看了看以前的教程。
salmon定量
## 定量获得TPM值
cd cleanData
mkdir ../salmon_output
index=$HOME/rna/SUPPA2/ref/gencode.v37.transcripts.salmon.index/
#index=$HOME/rna/SUPPA2/ref/gencode.v38.pc_transcripts.salmon.index
ls -lh $index
ls *gz|cut -d"_" -f 1|sort -u |while read id;do
nohup salmon quant -i $index -l A --gcBias \
-1 ${id}*_val_1.fq.gz -2 ${id}*_val_2.fq.gz -p 2 \
-o ../salmon_output/${id}_output 1>${id}_salmon.log 2>&1 &
done
## quant.sf文件很重要,要用于后续的分析
##ENST和ENSG的前三个字母(ENS),意思是“ENSENMBLE”。T是指转录本。G是指基因。当然,也会看到ENSP,P自然是指蛋白质了。
后续走 SUPPA2流程需要参考: https://mp.weixin.qq.com/s/kG2uI3me8Xo69mClThRutA
转录组流程结果文件夹结构
- qc
- raw
- fastqc,multiqc
- clean
- fastqc,multiqc
- trim
- txt
- align
- txt
- qualimap
- multiqc
- counts
- multiqc
- txt (based on name )
总表
mkdir qc && cd qc
mkdir raw_fq clean_fq align counts
nohup fastqc -t 10 ../raw/*gz -o raw_fq/ 1>log.rawFq.txt 2>&1 &
nohup fastqc -t 10 ../cleanData/*gz -o clean_fq/ 1>log.cleanFq.txt 2>&1 &
conda activate rna
multiqc -n clean_fq clean_fq/
感兴趣的小伙伴,可以去试试看做几个转录组项目哦:
ERR3640831
ERR3640832