突然间发现我们的RNA速率分析笔记仅仅是分享了在Linux操作的部分,见:使用基于python的velocyto软件做RNA速率分析,已经是2021年7月的事情了。
当时对很多测试项目都是拿到了loom文件,就可以进行下游 velocyto.R 这个R包进行后续统计可视化啦!准备第二天就讲解velocyto.R 这个R包用法,但是却忘记了,最近看到有小伙伴在该推文下面赞赏200元催更,激起了我的创作动力!
10x官网下载pbmc3k的bam文件走定量流程,我们提到了在10x官网下载pbmc3k数据集的bam文件,如下所示:
16G 6月 2 2017 pbmc3k_possorted_genome_bam.bam
然后使用bamtofastq命令把bam文件成功转为了fastq文件,自己看前面的笔记:10x官网下载pbmc3k的bam文件走定量流程,得到如下所示的文件:
$ ls -lh ../fastqs/gemgroup001/|cut -d" " -f 5- 1018M 3月 15 10:51 bamtofastq_S1_L000_I1_001.fastq.gz 1019M 3月 15 10:54 bamtofastq_S1_L000_I1_002.fastq.gz 1018M 3月 15 10:56 bamtofastq_S1_L000_I1_003.fastq.gz 591M 3月 15 10:58 bamtofastq_S1_L000_I1_004.fastq.gz 3.2G 3月 15 10:51 bamtofastq_S1_L000_R1_001.fastq.gz 3.2G 3月 15 10:54 bamtofastq_S1_L000_R1_002.fastq.gz 3.3G 3月 15 10:56 bamtofastq_S1_L000_R1_003.fastq.gz 2.0G 3月 15 10:58 bamtofastq_S1_L000_R1_004.fastq.gz 1.5G 3月 15 10:51 bamtofastq_S1_L000_R2_001.fastq.gz 1.5G 3月 15 10:54 bamtofastq_S1_L000_R2_002.fastq.gz 1.5G 3月 15 10:56 bamtofastq_S1_L000_R2_003.fastq.gz 861M 3月 15 10:58 bamtofastq_S1_L000_R2_004.fastq.gz 1.3G 3月 15 10:51 bamtofastq_S1_L000_R3_001.fastq.gz 1.3G 3月 15 10:54 bamtofastq_S1_L000_R3_002.fastq.gz 1.3G 3月 15 10:56 bamtofastq_S1_L000_R3_003.fastq.gz 768M 3月 15 10:58 bamtofastq_S1_L000_R3_004.fastq.gz
有了这个10x官网下载pbmc3k数据集的fastq文件,很容易重新走cellranger流程,得到新的bam文件,如下所示:
14G 3月 15 11:54 possorted_genome_bam.bam 4.1M 3月 15 11:55 possorted_genome_bam.bam.bai
可以看到,之前是16G的文件,现在成为了14G ,关于这个cellranger流程,就更简单了,我们也是在单细胞天地公众号详细介绍了cellranger全部使用细节及流程,大家可以自行前往学习,如下:
但是这个两年前的系列笔记是基于V2,V3版本的cellranger,在2020的7月我看到了其更新到了V4,也里面写了一个总结,见:cellranger更新到4啦(全新使用教程)
重新走pbmc3k数据集走RNA速率分析的上游流程
主要就是 bam2loom步骤,超级简单,见:使用基于python的velocyto软件做RNA速率分析,首先需要sort bam ,代码如下所示:
ls ./pbmc3k/outs/possorted_genome_bam.bam|while read id;do new=${id/possorted_genome_bam.bam/cellsorted_possorted_genome_bam.bam}
echo $new
nohup samtools sort -@ 10 -t CB -O BAM -o $new $id &
done
得到如下所示的文件:
15G 3月 15 12:07 cellsorted_possorted_genome_bam.bam
然后激活velocyto的小环境,运行velocyto这个Python程序即可:
conda activate velocyto
rmsk_gtf=$HOME/scRNA/msk/hg38_repeat_rmsk.gtf # 从genome.ucsc.edu下载
cellranger_gtf=/home/bakdata/x10/pipeline/refdata-gex-GRCh38-2020-A/genes/genes.gtf # 这个是cellranger官网提供的
nohup velocyto run10x -m $rmsk_gtf ./pbmc3k $cellranger_gtf &
运行结果在:
31M 3月 15 16:40 /pbmc3k/velocyto/pbmc3k.loom
假如是你自己的项目想follow我们的这个教程进行RNA速率分析,就必须要从bam文件开始啦,这样才有可能根据:使用基于python的velocyto软件做RNA速率分析得到loom文件。
下游是基于loom文件的velocyto.R可视化
直接读取pbmc3k的seurat对象文件和细胞注释信息,并且读取loom文件,进行两者的匹配。
首先是安装并且载入必备的包,代码如下所示:
rm(list = ls())
library(Seurat)
library(SeuratObject)
library(ggplot2)
# remotes::install_github('satijalab/seurat-wrappers')
library(SeuratWrappers) #RunVelocity
# library(devtools)
# remotes::install_github("velocyto-team/velocyto.R")
library(velocyto.R)
library(stringr)
然后以大家熟知的pbmc3k数据集为例,读取pbmc3k的seurat对象结果,大家先安装这个数据集对应的包,并且对它进行降维聚类分群,参考前面的例子:人人都能学会的单细胞聚类分群注释 ,而且每个亚群找高表达量基因,都存储为Rdata文件。
library(SeuratData) #加载seurat数据集
getOption('timeout')
options(timeout=10000)
#InstallData("pbmc3k")
data("pbmc3k")
sce <- pbmc3k.final
library(Seurat)
table(Idents(sce))
p1=DimPlot(sce,label = T)
sce$celltype = Idents(sce)
DimPlot(sce,reduction = "umap",label=T,label.size = 6)+theme_bw()
因为这个数据集已经进行了不同单细胞亚群的标记,所以我们很容易拿到单细胞表达量矩阵和细胞对应的表型信息。可视化如下所示:
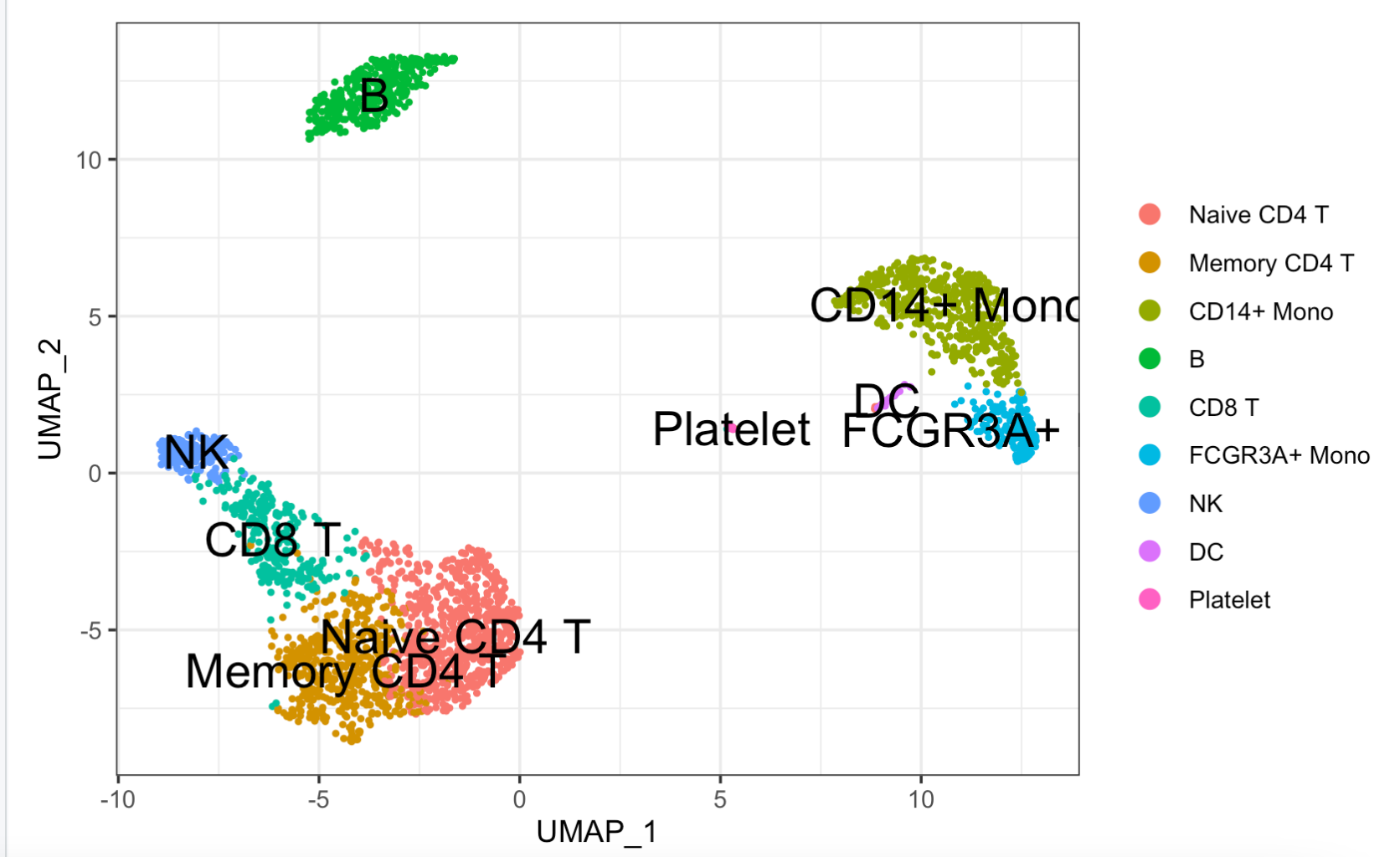
######读取loom文件
ldat <- read.loom.matrices("./pbmc3k.loom")
DefaultAssay(sce) <- 'RNA'
#查看loom文件和seurat的细胞与基因名
ldat$spliced[1:3,1:3]
sce@meta.data[1:3,1:3]
####根据seurat的barcodes修改loom文件的barcodes
colnames(ldat$spliced) <- gsub("x","",gsub(":","_",colnames(ldat$spliced)))
head(colnames(ldat$spliced) )
colnames(ldat$spliced) <- gsub("pbmc3k_","", colnames(ldat$spliced) )
colnames(ldat$unspliced) <- colnames(ldat$spliced)
colnames(ldat$ambiguous) <- colnames(ldat$spliced)
head(rownames(sce@meta.data))
head( colnames(ldat$spliced))
需要自己肉眼看两个对象的细胞名字是匹配的哦,每次都需要耗费很多时间去仔细看。
这里我们仅仅是关心单核细胞的关系,就是筛选一下seurat对象,代码如下所示:
table(sce$celltype)
# sce2 = sce[,sce$celltype %in% c( 'CD14-Mono' , 'CD16-Mono')]
sce2 = sce[,sce$celltype %in% c('CD14+ Mono' , 'FCGR3A+ Mono')]
table(sce2$celltype)
DimPlot(sce2,reduction = "umap",label=T,label.size = 6) +theme_bw()
## 由于Seurat的对象筛选了数据,所以两个文件细胞并不相同,以Seurat对象为准
ldat$spliced<-ldat$spliced[,rownames(sce2@meta.data)]
ldat$unspliced<-ldat$unspliced[,rownames(sce2@meta.data)]
ldat$ambiguous<-ldat$ambiguous[,rownames(sce2@meta.data)]
sp <- ldat$spliced
unsp <- ldat$unspliced
#提取seurat对象的降维umap坐标
umap <- sce2@reductions$umap@cell.embeddings
#将loom转换为seurat
bm <- as.Seurat(x = ldat)
#将降维信息赋给bm
bm@reductions <- sce2@reductions
#将meta信息赋给bm
bm@meta.data <- sce2@meta.data
table(bm$celltype)
Idents(bm) <- bm$celltype
可以看到, 我们把 loom文件里面的信息,转为了seurat对象哦,而且直接复制了pbmc3k数据集的降维聚类分群坐标信息,无需对loom文件进行降维聚类分群啦。接下来就对这个loom文件的seurat对象进行 RunVelocity 操作:
## 估计细胞和细胞的距离
cell.dist <- as.dist(1-armaCor(t(sce2@reductions$umap@cell.embeddings)))
fit.quantile <- 0.02
bm <- RunVelocity(object = bm,
deltaT = 1, kCells = 25,
fit.quantile = 0.02,cell.dist=cell.dist)
ident.colors <- (scales::hue_pal())(n = length(x = levels(x = bm)))
names(x = ident.colors) <- levels(x = bm)
cell.colors <- ident.colors[Idents(object = bm)]
names(x = cell.colors) <- colnames(x = bm)
RunVelocity 操作后就是简单的可视化啦:
emb = Embeddings(object = bm, reduction = "umap")
show.velocity.on.embedding.cor(emb, vel = Tool(object = bm, slot = "RunVelocity"),
n=200,scale='sqrt',
cell.colors=ac(cell.colors,alpha=0.5),cex=0.8,
arrow.scale=3,show.grid.flow=TRUE,
min.grid.cell.mass=0.5,grid.n=40,arrow.lwd=1,
do.par=F,cell.border.alpha = 0.1)
参数有点多,感兴趣的可以自行细致的解读, 直接让我们看结果吧:
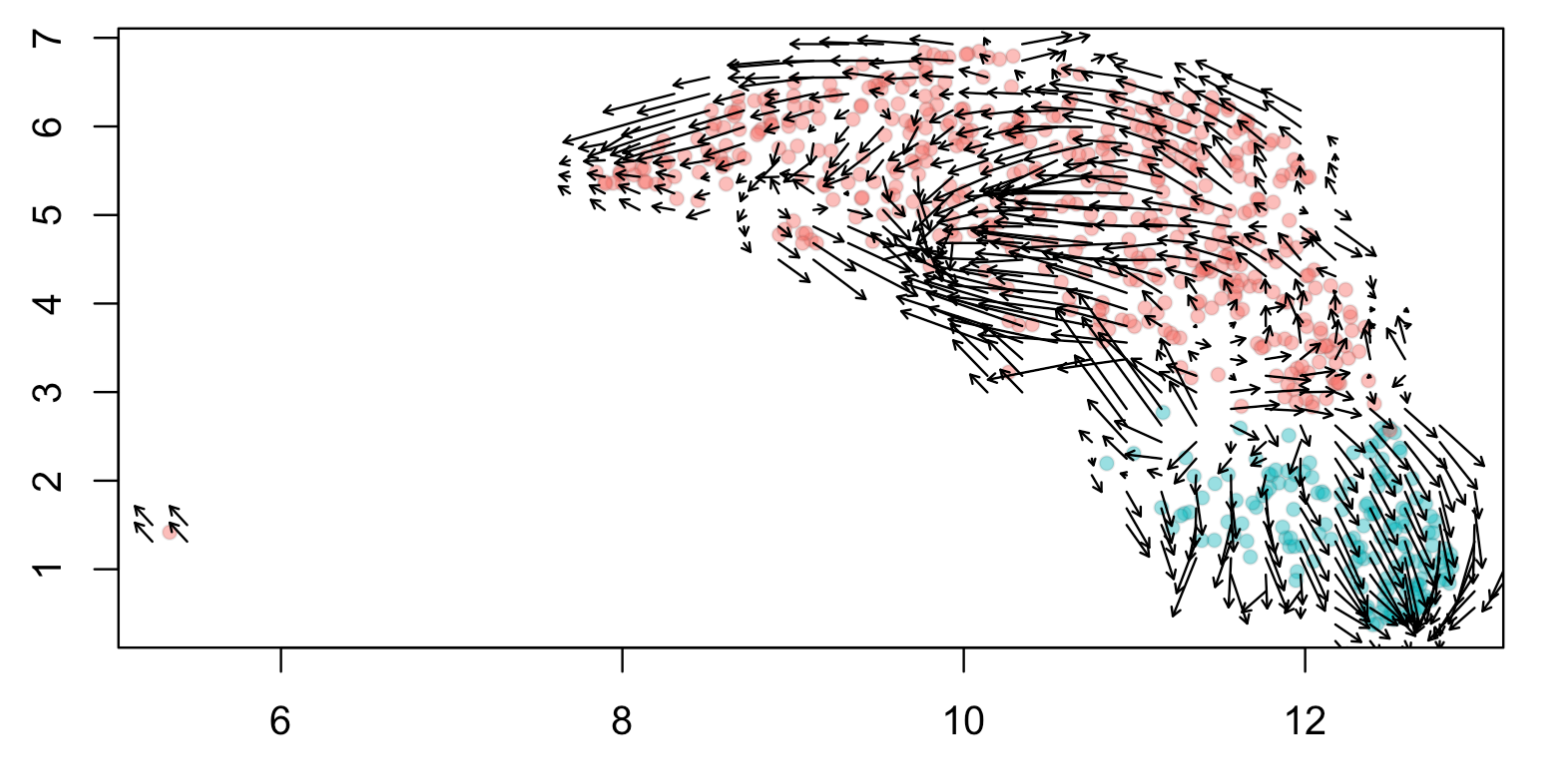
可以看到 CD14+ Mono 和 FCGR3A+ Mono 从RNA速率的角度来看,并不是一个线性的 关系。可以跟我们前面的拟时序分析的结果对比,见:拟时序分析就是差异分析的细节剖析,提取了 CD14+ Mono 和 FCGR3A+ Mono这两个单细胞亚群,看其细节的变化,但是因为monocle其实得到的pseudotime值是没有方向,大小的顺序是可以调整的, 所以我们仅仅是凭借下面的拟时序图表没办法判断CD14+ Mono 和 FCGR3A+ Mono的分化关系:
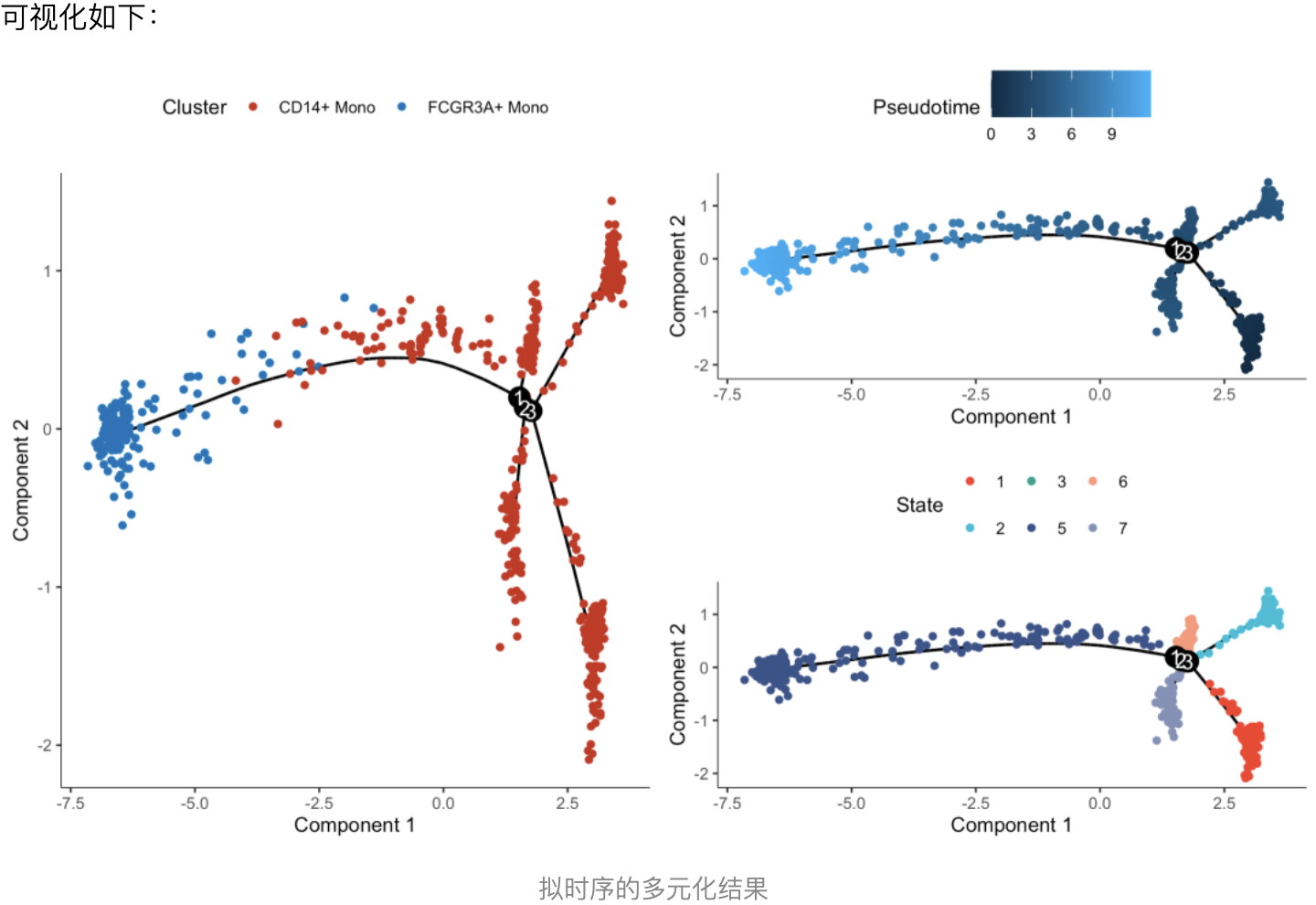
从拟时序的角度其实只能是看到 CD14+ Mono 和 FCGR3A+ Mono 非常的不一样,所以它们在拟时序图上面泾渭分明,而且呢,这个 CD14+ Mono 的内部异质性非常大,从上面的RNA速率的角度来看,确实是FCGR3A+ Mono 的箭头纯粹一点,而CD14+ Mono 的内部异质性大所以里面的RNA速率的箭头也是多个终点。
大家需要自己根据前面的笔记:10x官网下载pbmc3k的bam文件走定量流程,以及:使用基于python的velocyto软件做RNA速率分析得到这个pbmc3k数据集的loom文件哦。
然后才能进行我们上面的velocyto.R可视化,这些代码都有一点门槛,即需要Linux又需要R语言。
再怎么强调生物信息学数据分析学习过程的计算机基础知识的打磨都不为过,我把它粗略的分成基于R语言的统计可视化,以及基于Linux的NGS数据处理:
把R的知识点路线图搞定,如下:
-
了解常量和变量概念
-
加减乘除等运算(计算器)
-
多种数据类型(数值,字符,逻辑,因子)
-
多种数据结构(向量,矩阵,数组,数据框,列表)
-
文件读取和写出
-
简单统计可视化
-
无限量函数学习
Linux的6个阶段也跨越过去 ,一般来说,每个阶段都需要至少一天以上的学习:
-
第1阶段:把linux系统玩得跟Windows或者MacOS那样的桌面操作系统一样顺畅,主要目的就是去可视化,熟悉黑白命令行界面,可以仅仅以键盘交互模式完成常规文件夹及文件管理工作。
-
第2阶段:做到文本文件的表格化处理,类似于以键盘交互模式完成Excel表格的排序、计数、筛选、去冗余、查找、切割、替换、合并、补齐,熟练掌握awk、sed、grep这文本处理的三驾马车。
-
第3阶段:元字符,通配符及shell中的各种扩展,从此linux操作不再神秘!
-
第4阶段:高级目录管理:软硬链接,绝对路径和相对路径,环境变量。
-
第5阶段:任务提交及批处理,脚本编写解放你的双手。
-
第6阶段:软件安装及conda管理,让linux系统实用性放飞自我。