因为众所周知的原因(详见: 很抱歉我这里没有朝九晚五 ,以及得了吧,不缺你这点关注 ),我现在是没有实习生了。但是我还有学徒,所以可以在培养 他们的同时,给他们布置一些数据分析实战任务。
这次我给学徒布置了一个非小细胞肺癌的免疫治疗的单细胞转录组数据,文章是2023的《Tumor microenvironment remodeling after neoadjuvant immunotherapy in non-small cell lung cancer revealed by single-cell RNA sequencing》,因为我注意到里面的降维聚类分群有大量的中性粒细胞,如下所示:
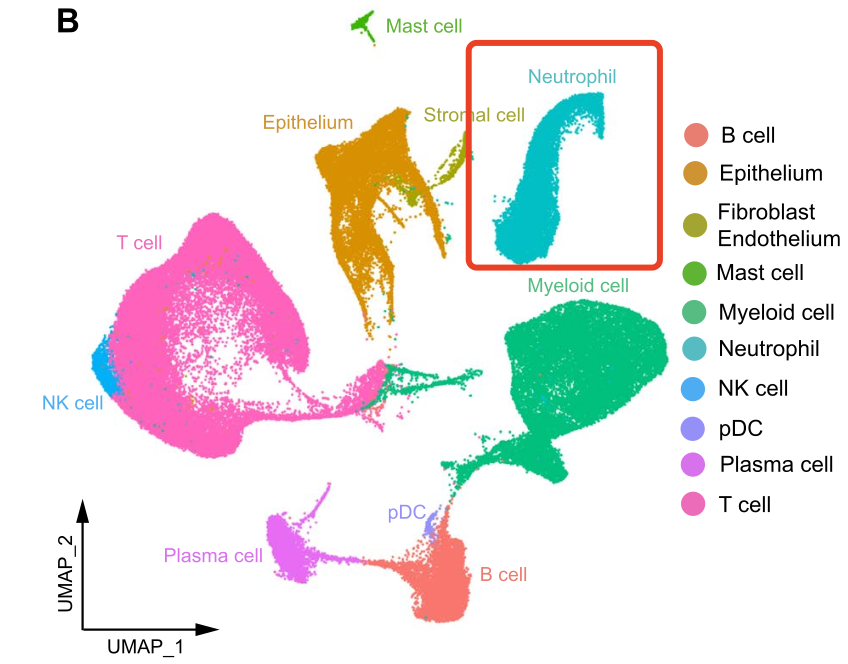
这个中性粒细胞是BD平台的特色,很容易检索到:
而且它的数据是公开的,在GSE207422,链接是;https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE207422
Supplementary file Size Download File type/resource
GSE207422_NSCLC_bulk_RNAseq_log2TPM.txt.gz 5.4 Mb (ftp)(http) TXT
GSE207422_NSCLC_bulk_RNAseq_metadata.xlsx 11.8 Kb (ftp)(http) XLSX
GSE207422_NSCLC_scRNAseq_UMI_matrix.txt.gz 175.5 Mb (ftp)(http) TXT
GSE207422_NSCLC_scRNAseq_metadata.xlsx 11.1 Kb (ftp)(http) XLSX
有了上面的文件,很容易根据我教大家的单细胞转录组数据分析流程来完成上面的图表复现。但是我拿到了学徒给我的结果后就傻眼了,完全是看不到中性粒细胞啊, 也就是说BD平台单细胞的优点都被你弄丢了。。。
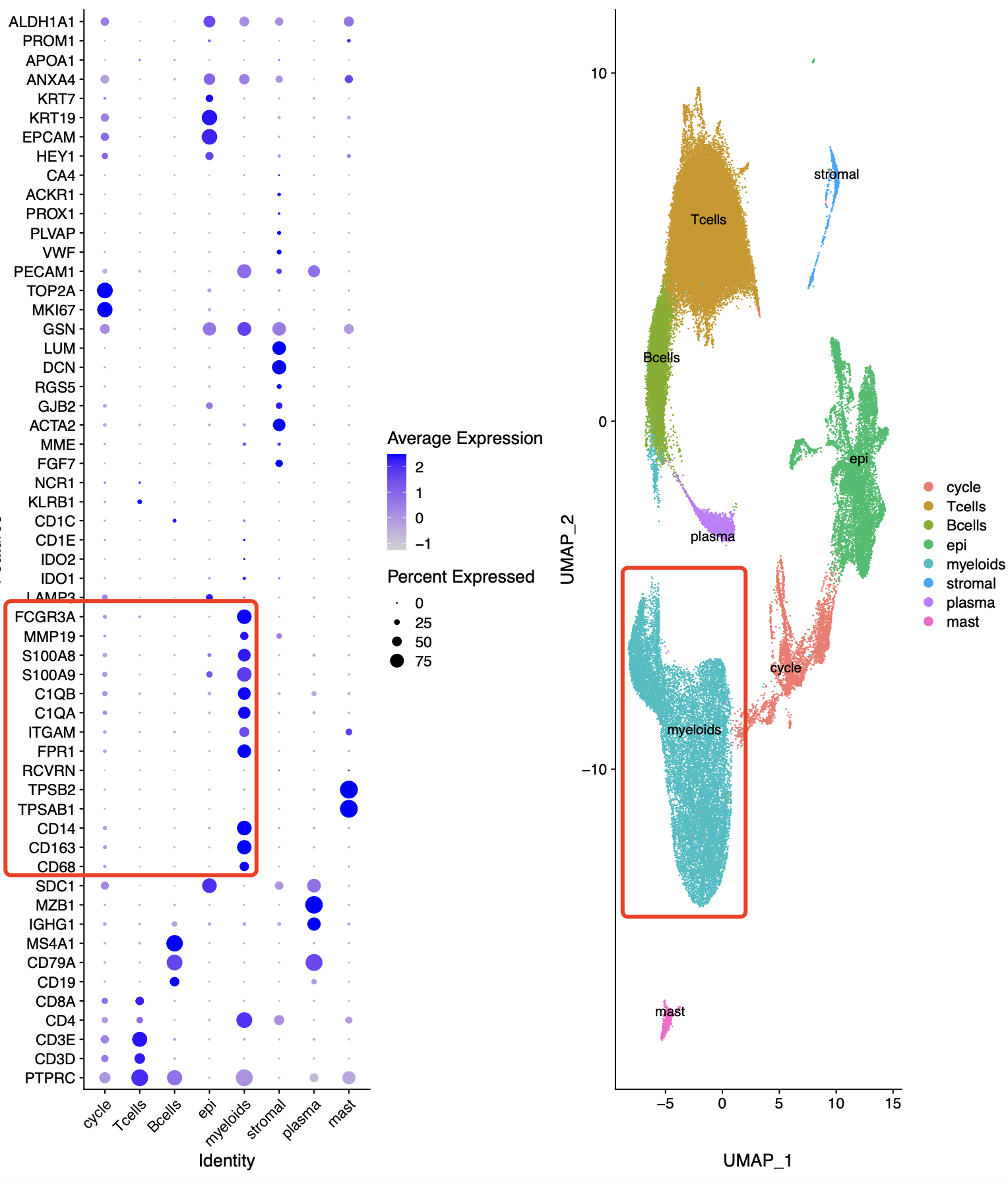
通常我们拿到了肿瘤相关的单细胞转录组的表达量矩阵后的第一层次降维聚类分群通常是:
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibo or CD31+,PECAM1,endo)
参考我前面介绍过 CNS图表复现08—肿瘤单细胞数据第一次分群通用规则,这3大单细胞亚群构成了肿瘤免疫微环境的复杂。绝大部分文章都是抓住免疫细胞亚群进行细分,包括淋巴系(T,B,NK细胞)和髓系(单核,树突,巨噬,粒细胞)的两大类作为第二次细分亚群。但是也有不少文章是抓住stromal 里面的fibo 和endo进行细分,并且编造生物学故事的。
我们前面已经对免疫细胞里面的髓系和B细胞细分亚群进行了简单的介绍:
但是之前的髓系免疫细胞细分的时候其实并没有中性粒细胞亚群,它也一直不在我们分享的单细胞数据处理代码里面。这次既然是学徒失败了,那我们就一起看看是不是学徒的哪个步骤有问题。。。。
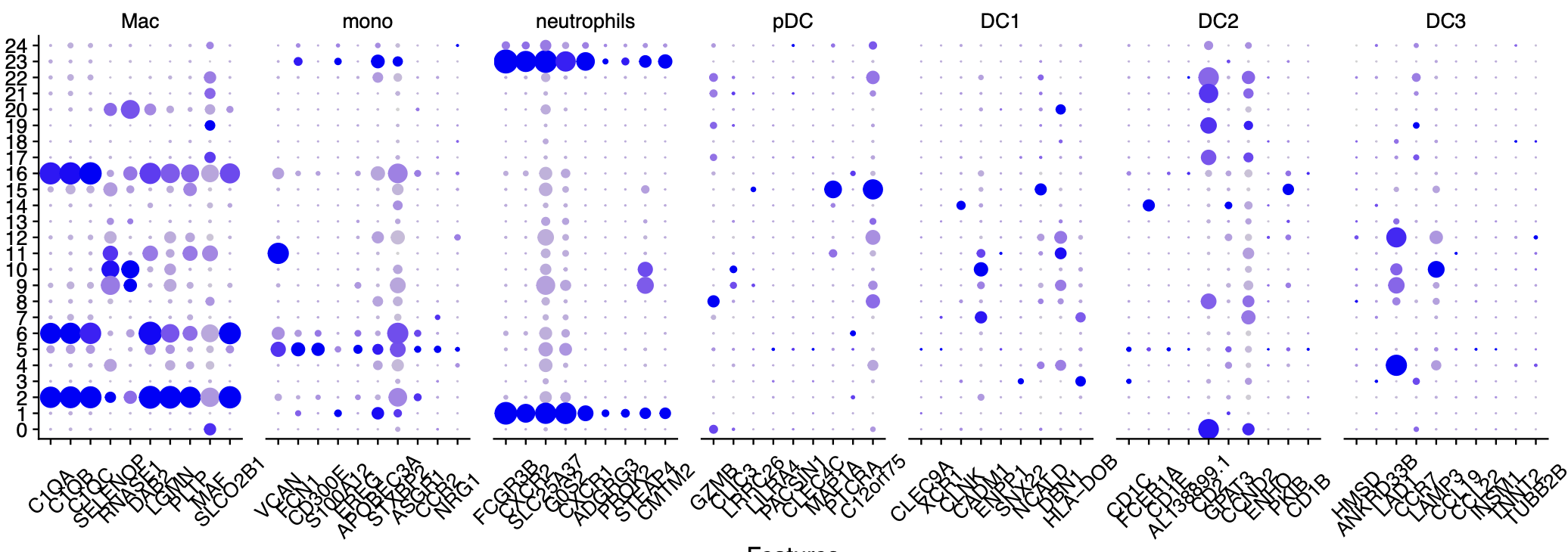
首先查看中性粒细胞的特异性基因
我把分辨率调很高,发现也是没有中性粒细胞的特异性基因的高表达亚群,如下所示:
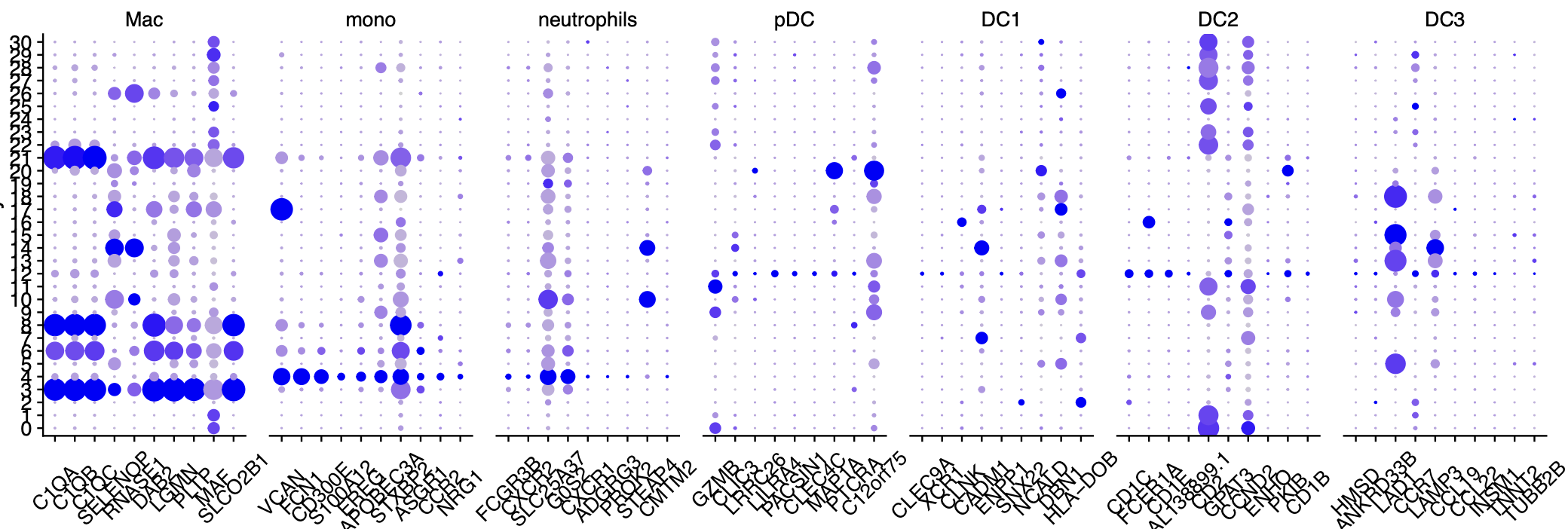
如果要查看中性粒细胞的特异性基因代码也很简单:
th=theme(axis.text.x = element_text(angle = 45,
vjust = 0.5, hjust=0.5))
myeloids = list(
Mac=c("C1QA","C1QB","C1QC","SELENOP","RNASE1","DAB2","LGMN","PLTP","MAF","SLCO2B1"),
mono=c("VCAN","FCN1","CD300E","S100A12","EREG","APOBEC3A","STXBP2","ASGR1","CCR2","NRG1"),
neutrophils = c("FCGR3B","CXCR2","SLC25A37","G0S2","CXCR1","ADGRG3","PROK2","STEAP4","CMTM2" ),
pDC = c("GZMB","SCT","CLIC3","LRRC26","LILRA4","PACSIN1","CLEC4C","MAP1A","PTCRA","C12orf75"),
DC1 = c("CLEC9A","XCR1","CLNK","CADM1","ENPP1","SNX22","NCALD","DBN1","HLA-DOB","PPY"),
DC2=c( "CD1C","FCER1A","CD1E","AL138899.1","CD2","GPAT3","CCND2","ENHO","PKIB","CD1B"),
DC3 = c("HMSD","ANKRD33B","LAD1","CCR7","LAMP3","CCL19","CCL22","INSM1","TNNT2","TUBB2B")
)
p <- DotPlot(sce.all, features = myeloids,
assay='RNA' ,group.by = 'celltype' ) +th
p
ggsave(plot=p, filename="check_myeloids_marker_by_celltype.pdf",width = 15)
p <- DotPlot(sce.all, features = myeloids,
assay='RNA' ,group.by = 'RNA_snn_res.0.8' ) +th
p
ggsave(plot=p, filename="check_myeloids_marker_by_RNA_snn_res.0.8.pdf",
width = 15)
可以看到,我使用了 RNA_snn_res.0.8 这样的分辨率,仍然是看不到特异性中性粒细胞亚群。
这就让我很诧异,前些天就遇到了:BD单细胞转录组分析怕不是一个笑话吧,是那个数据集GSE201088的文章作者自己把bd单细胞转录组数据分析做错了,并不是bd单细胞本身问题, 这次难道又是文章作者问题?
但是我细看2023的《Tumor microenvironment remodeling after neoadjuvant immunotherapy in non-small cell lung cancer revealed by single-cell RNA sequencing》,文章里面其实是有数量不少的中性粒细胞啊,那就是学徒自己的数据处理问题。但是学徒使用的是我的标准代码,这个代码我处理了市面上一千多个单细胞公共数据集,没有遇到问题,这次怎么就不对了呢?
我就帮学徒打开我的代码调试了一下,发现出现在过滤参数,文章描述的有如下所示的 过滤:

可以看到,里面其实是并没有核糖体的阈值,而我们的标准代码里面:
#过滤指标2:线粒体/核糖体基因比例(根据上面的violin图)
selected_mito <- WhichCells(input_sce.filt, expression = percent_mito < 25)
selected_ribo <- WhichCells(input_sce.filt, expression = percent_ribo > 3)
selected_hb <- WhichCells(input_sce.filt, expression = percent_hb < 1 )
只需要我注释掉这个核糖体过滤的代码,接下来的降维聚类分群就正常了,如下所示:
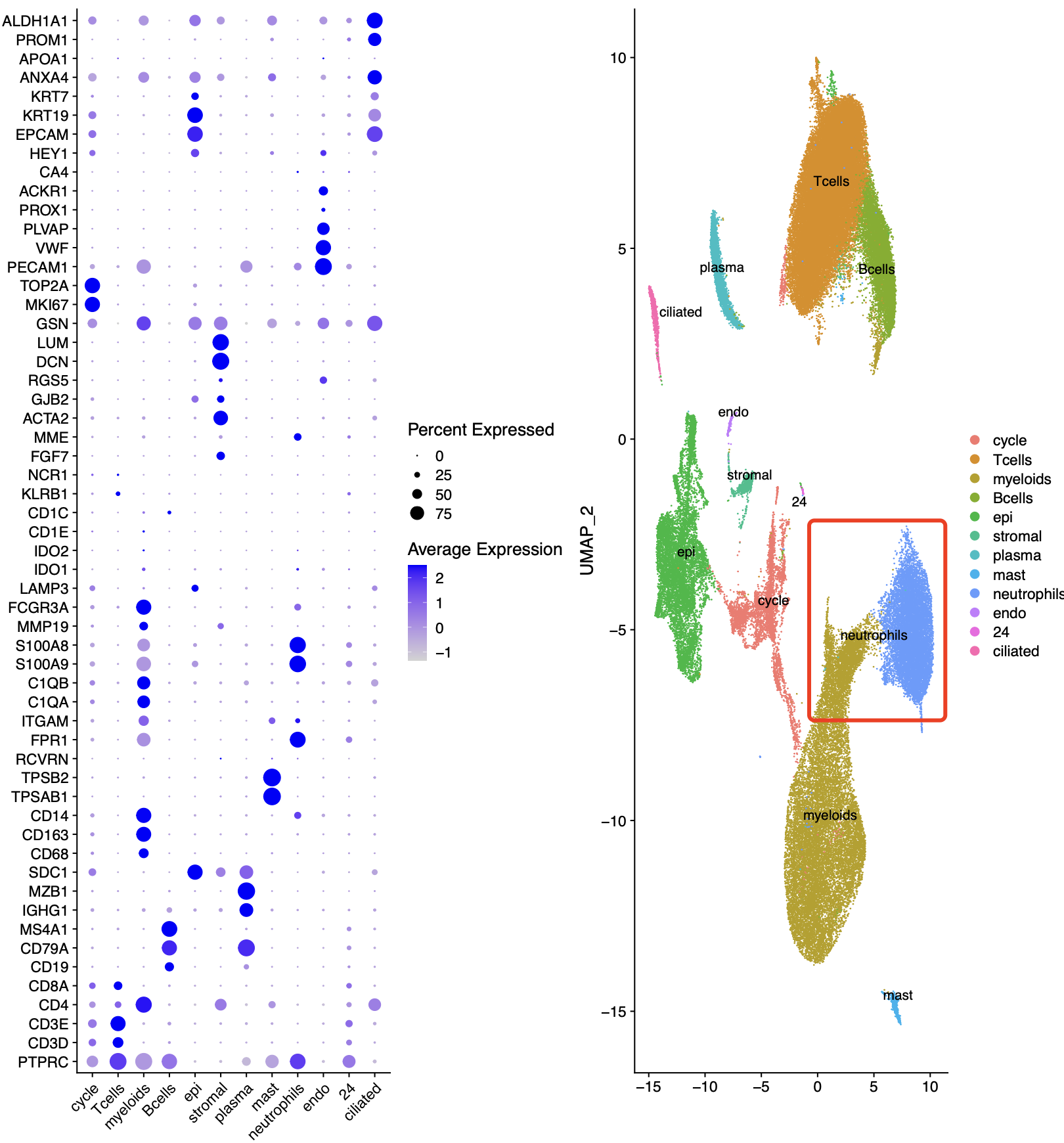
可以看到是非常清晰的中性粒细胞的特异性基因的高表达亚群,如下所示:
仍然是手动定义了单细胞亚群的生物学名字:
#定义细胞亚群
celltype[celltype$ClusterID %in% c( 0,17,19,21 ),2]='Tcells'
celltype[celltype$ClusterID %in% c( 3 ),2]='Bcells'
celltype[celltype$ClusterID %in% c( 7 ),2]='plasma'
celltype[celltype$ClusterID %in% c( 2,5,6 ),2]= 'myeloids' #
celltype[celltype$ClusterID %in% c( 1,23),2]='neutrophils'
celltype[celltype$ClusterID %in% c( 14),2]='mast'
celltype[celltype$ClusterID %in% c( 11 ),2]='stromal'
celltype[celltype$ClusterID %in% c( 20 ),2]='endo'
celltype[celltype$ClusterID %in% c(8,12,16,22),2]='cycle'
初学者不仔细就容易遇到各种各样的问题
我们五年前就系统性整理了全部的单细胞转录组数据分析代码,它理论上是可以应用于几乎所有的单细胞数据集,因为几乎所有的单细胞转录组数据都来源于10x单细胞平台,但是这样的bd单细胞平台,就需要一定程度的自定义了。。。。
核心代码如下所示:
##
### ---------------
###
### Create: Jianming Zeng
### Date: 2023-01-16 20:53:22
### Email: jmzeng1314@163.com
### Blog: http://www.bio-info-trainee.com/
### Forum: http://www.biotrainee.com/thread-1376-1-1.html
### CAFS/SUSTC/Eli Lilly/University of Macau
### Update Log: 2023-01-16 First version
###
### ---------------
rm(list=ls())
options(stringsAsFactors = F)
source('scRNA_scripts/lib.R')
library(readxl)
###### step1:导入数据 ######
# 付费环节 800 元人民币
ct=fread( 'GSE207422_RAW/GSE207422_NSCLC_scRNAseq_UMI_matrix.txt.gz',
data.table = F)
ct[1:4,1:4]
ct[nrow(ct),1:4]
rownames(ct)=ct[,1]
ct=ct[,-1]
ct[1:4,1:4]
ct1 = ct
s <- colnames(ct1)
s[1:10]
s1 = substr(s, 1, 11)
s1[1:10]
table(s1)
colnames(ct1) = s1
dim(ct1)
# [1] 24292 92330
# 这个时候的细胞数量跟文章一致
# After quality control and removal of doublets,
# transcriptomes from 92,330 cells with a median of 1256 genes per cell
# were used for further analyses.
meta <- read_excel("GSE207422_RAW/GSE207422_NSCLC_scRNAseq_metadata.xlsx")
sce.all = CreateSeuratObject(counts = ct1 ,
min.cells = 5,
min.features = 500)
sce.all
as.data.frame(sce.all@assays$RNA@counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all@meta.data$orig.ident)
s = colnames(sce.all)
s1 = substr(s, 1, 11)
s1[1:10]
table(s1)
sce.all$orig.ident = s1
table(sce.all$orig.ident )
s1 = gsub(c('BD_immune01|BD_immune05|BD_immune08'),"Pre-treatment",s1)
table(s1)
s2 = gsub("BD_immune.*$", "Post-treatment", s1) #第一个空格直达结尾替换成b
table(s2)
phe=str_split( colnames(sce.all) ,'_',simplify = T)
head(phe)
table(phe[,1])
sce.all$group = s2
head(sce.all@meta.data)
table(sce.all$group)
table(sce.all$orig.ident)
gplots::balloonplot(table(sce.all$group,sce.all$orig.ident))
###### step2:QC质控 ######
dir.create("./1-QC")
setwd("./1-QC")
# 如果过滤的太狠,就需要去修改这个过滤代码
source('../scRNA_scripts/qc.R')
sce.all.filt = basic_qc(sce.all)
print(dim(sce.all))
print(dim(sce.all.filt))
setwd('../')
sp='human'
###### step3: harmony整合多个单细胞样品 ######
dir.create("2-harmony")
getwd()
setwd("2-harmony")
source('../scRNA_scripts/harmony.R')
# 默认 ScaleData 没有添加"nCount_RNA", "nFeature_RNA"
# 默认的
sce.all.int = run_harmony(sce.all.filt)
setwd('../')
###### step4: 降维聚类分群和看标记基因库 ######
# 原则上分辨率是需要自己肉眼判断,取决于个人经验
# 为了省力,我们直接看 0.1和0.8即可
table(Idents(sce.all.int))
table(sce.all.int$seurat_clusters)
table(sce.all.int$RNA_snn_res.0.1)
table(sce.all.int$RNA_snn_res.0.8)
getwd()
dir.create('check-by-0.1')
setwd('check-by-0.1')
sel.clust = "RNA_snn_res.0.1"
sce.all.int <- SetIdent(sce.all.int, value = sel.clust)
table(sce.all.int@active.ident)
source('../scRNA_scripts/check-all-markers.R')
setwd('../')
getwd()
dir.create('check-by-0.8')
setwd('check-by-0.8')
sel.clust = "RNA_snn_res.0.8"
sce.all.int <- SetIdent(sce.all.int, value = sel.clust)
table(sce.all.int@active.ident)
source('../scRNA_scripts/check-all-markers.R')
setwd('../')
getwd()
last_markers_to_check
上面的代码需要用到 scRNA_scripts 文件夹里面的几个R脚本,它已经被我运用到一千多个单细胞公共数据集,没有遇到问题!但是,最近Seurat包出现了V5的更新了,而我前面的代码都是V4,就尴尬了!!!