虽然我们一再强调:假如你不喜欢最新版的Seurat包的单细胞理念,大家完全是可以选择降级这个Seurat。主要是因为很多初学者拿到了大量的基于V4版本Seurat的教程会手足无措,其实很容易迁移。所以我们也在学员们的催促下转向了Seurat的V5版本,详见:从零开始配置R编程语言软件环境,而且是在视频号有直播回放,详见:
虽然说我们安装了Seurat的V5版本,但是初次使用的时候加载就报错了,如下所示:
The sp package is now running under evolution status 2
(status 2 uses the sf package in place of rgdal)
Error: package or namespace load failed for ‘SeuratObject’ in loadNamespace(j <- i[[1L]], c(lib.loc, .libPaths()), versionCheck = vI[[j]]):
载入了名字空间‘Matrix’ 1.6-1,但需要的是>= 1.6.3
错误: 无法载入程辑包‘SeuratObject’
很明显是Matrix版本问题,只需要卸载它后,更新一下即可,如下所示:
> remove.packages("Matrix")
从‘C:/Users/jimmy/AppData/Local/R/win-library/4.3’中删除程序包
(因为没有指定‘lib’)
> install.packages('Matrix')
将程序包安装入‘C:/Users/jimmy/AppData/Local/R/win-library/4.3’
(因为‘lib’没有被指定)
试开URL’https://cran.rstudio.com/bin/windows/contrib/4.3/Matrix_1.6-4.zip'
Content type 'application/zip' length 4562987 bytes (4.4 MB)
downloaded 4.4 MB
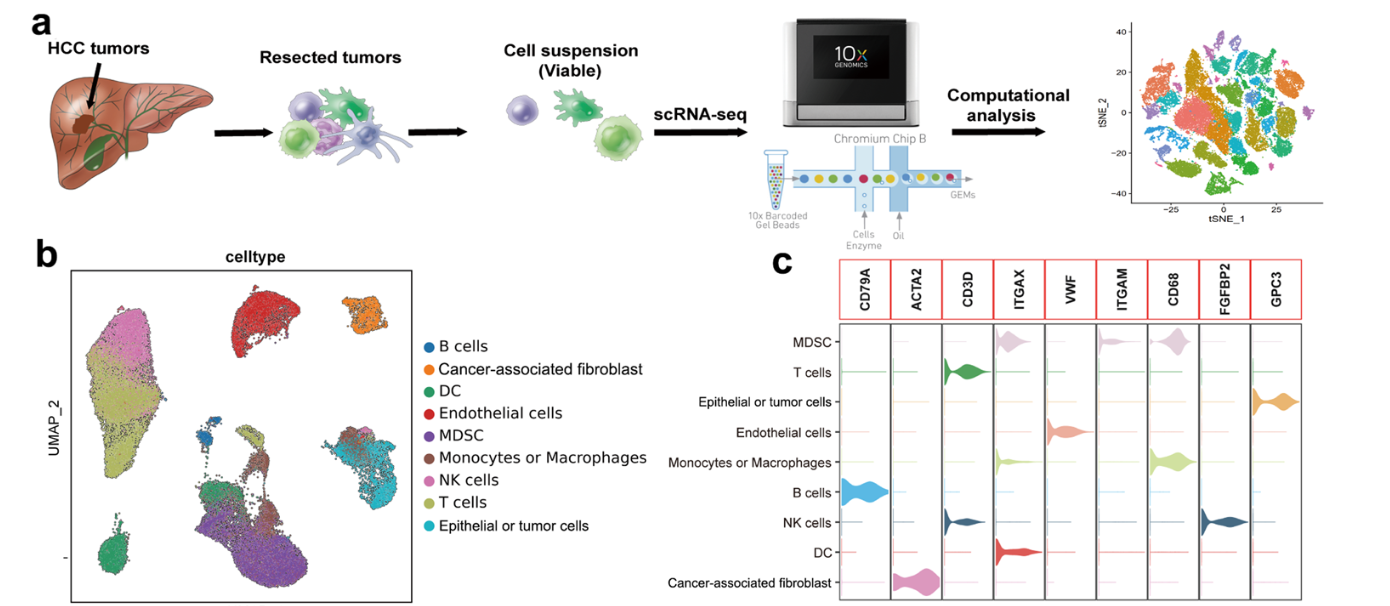
接下来我们就读取10X文件的3个标准文件,使用如下所示的例子,大家可以去自己下载 (2023-GSE202642-肝癌-成纤维单细胞亚群),文章标题是:《CD36+ cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor》,里面的降维聚类分群如下所示:
在 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE202642 可以看到作者给出来的3个文件的表达量矩阵:
GSE202642_barcodes.tsv.gz 569.6 Kb (ftp)(http) TSV
GSE202642_features.tsv.gz 325.6 Kb (ftp)(http) TSV
GSE202642_matrix.mtx.gz 666.5 Mb (ftp)(http) MTX
如下所示:
这个时候使用Seurat的V5版本和之前Seurat的V4版本读取方式并没有本质上区别,都是:
sce.all=CreateSeuratObject(counts = Read10X( 'GSE202642/' ) ,
min.cells = 5,
min.features = 300,)
library(stringr)
sce=sce.all
head(rownames(sce@meta.data))
phe=str_split(rownames(sce@meta.data),'-',simplify = T)
head(phe)
tail(phe)
table(phe[,2])
sce@meta.data$orig.ident=paste0('p',phe[,2])
table(sce@meta.data$orig.ident)
读取之后我就简单的检查一下这个Seurat对象里面的信息,然后遇到第一个报错
> as.data.frame(sce.all@assays$RNA@counts[1:10, 1:2])
Error in as.data.frame(sce.all@assays$RNA@counts[1:10, 1:2]) :
"counts"槽名不存在于"Assay5"类别对象中
其实是很简单的debug,只需要查看一下这个Seurat对象结构,就知道了:
as.data.frame(sce.all@assays$RNA@counts[1:10, 1:2])
# 上面的错误
# 可以修改为下面的两种
as.data.frame(sce.all@assays$RNA@layers$counts[1:10, 1:2])
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
真是搞不懂,为什么这么简单的bug就让大家放弃了Seurat的V5,简直是滑天下之大稽。这么多人学了这么久的R代码就之后照抄我的案例代码吗,不会活学活用吗?
而且,这个bug根本就并不会影响整个Seurat数据分析流程啊, 降维聚类分群仍然是ok的。