前面我们在推文:数据分析有错误并不可怕,造假才不可饶恕 提到了这个新鲜出炉( 2023年12月5日)的cell期刊的文章单细胞转录组数据分析环节有一些值得探讨的地方,比如第一层次降维聚类分群后的亚群的生物学命名,就发现很多特异性高表达的基因并不主流,而且很多主流基因是缺失的。
更麻烦的是因为文献里面的两个分组每个组内都是3个样品而已,而文章大家结论缺依赖于这个单细胞水平的细胞比例变化。其实早期(2018-2021时候)单细胞转录组费用居高不下,所以绝大部分情况下大家做两个分组,每个组内也就是三五个样品而已。这样的话两个分组之间的不同单细胞亚群的比例差异其实往往是需要最后使用流式细胞等价格相对低廉的实验技术去扩大样品队列去验证一下。
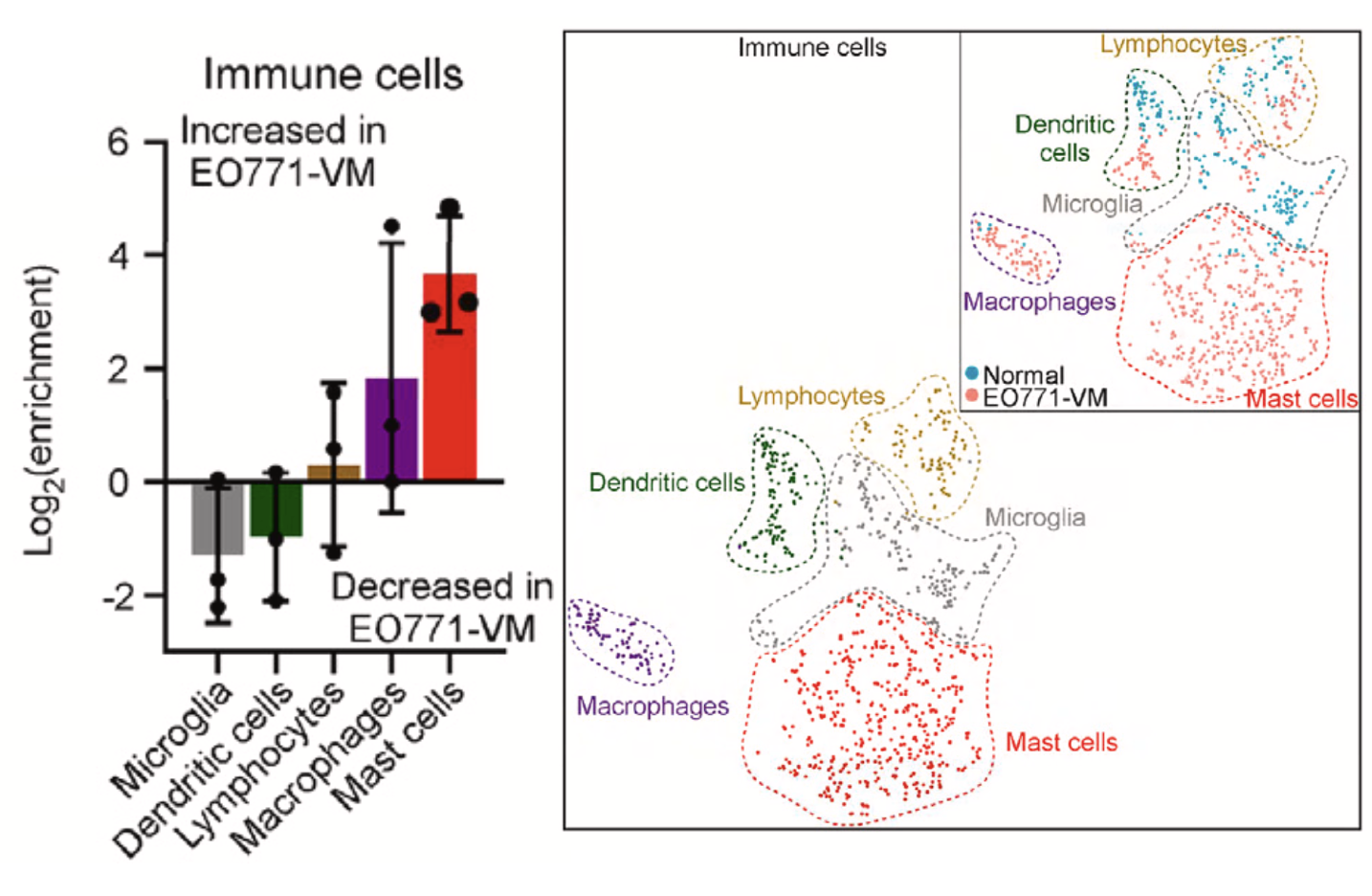
肉眼可以看到免疫细胞比例变化
如下所示,可以看到免疫细胞主要是来源于处理组,两个分组各自的单细胞数量分别是:
- 10,443 nuclei from normal mice
- 7,846 nuclei from EO771-VM-bearing mice
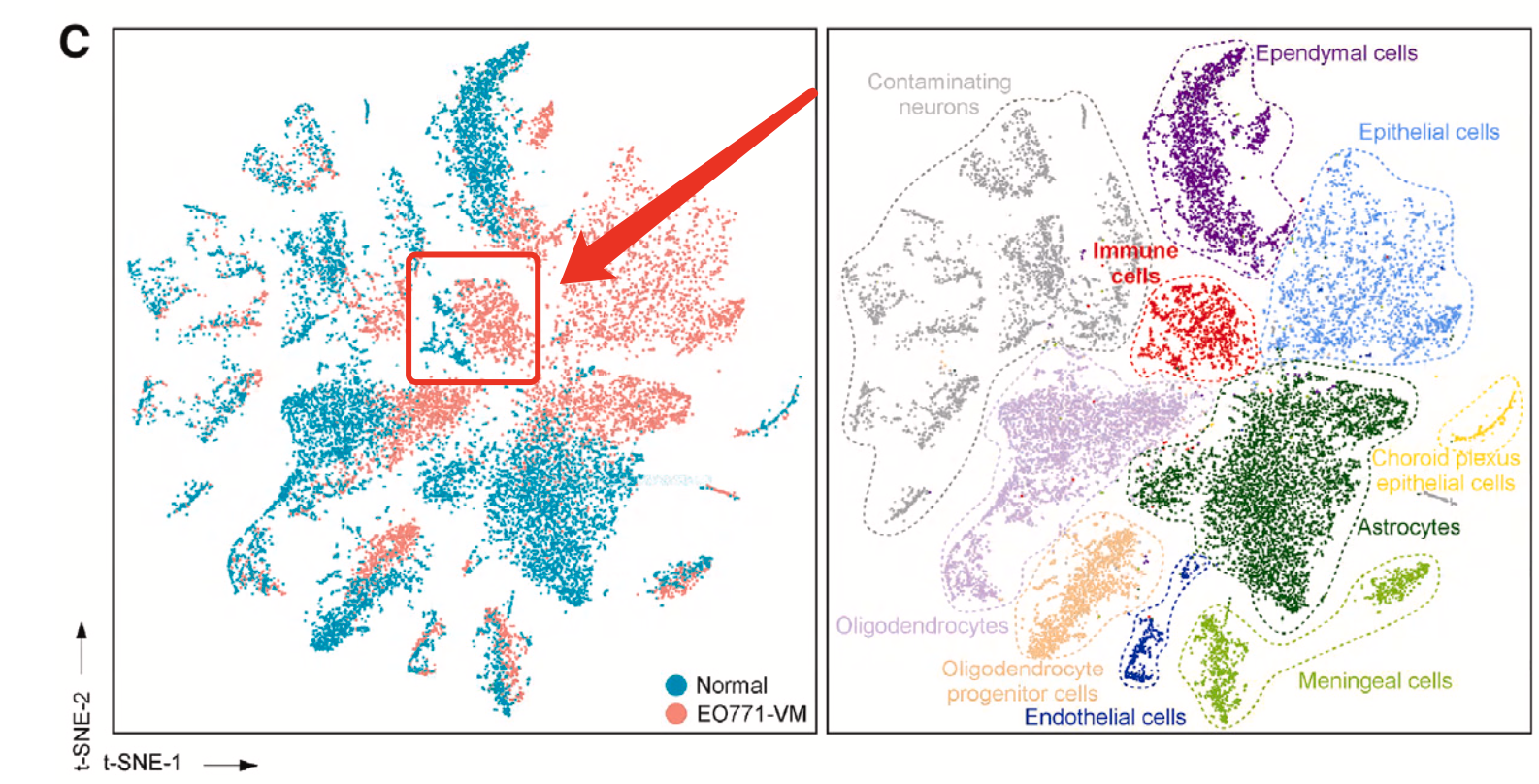
虽然说从上面的二维的t-distributed sto- chastic neighbor embedding (t-SNE) 的散点图是可以看出来在处理组(EO771-VM-bearing mice)的免疫细胞绝对数量是远多于正常组,但是作者仍然是采用了下面的比例图来从另外一个维度说明处理组的免疫细胞相对数量是远多于正常组,如下所示的条形图:
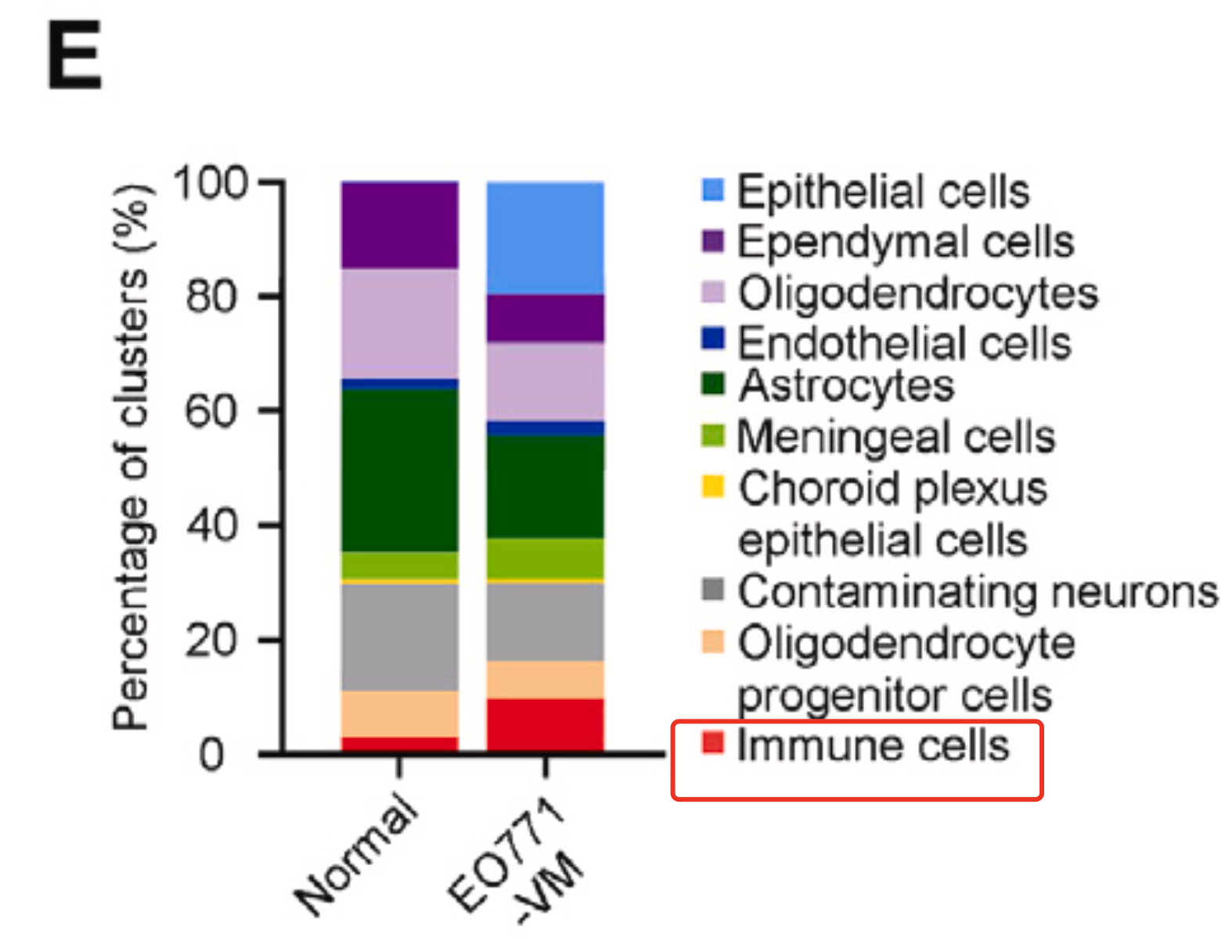
当然了,上面的图里面的比例变化最夸张的当然是恶性的肿瘤上皮细胞啦,因为脑转移成功样品里面的才会有恶性的肿瘤上皮细胞,文献里面是这样描述这个模型的: - mouse breast cancer EO771 cells and Lewis lung carcinoma (LLC) cells were injected into the internal carotid artery of syngeneic C57BL/6 mice,
- while human breast cancer MDA-MB-231 cells were inoculated into nude mice.
绝对数量和相对数量就足够了吗
我们在前面的推文:数据分析有错误并不可怕,造假才不可饶恕 给出来了代码,大家简单的复制粘贴即可运行第一层次降维聚类分群,如下所示:
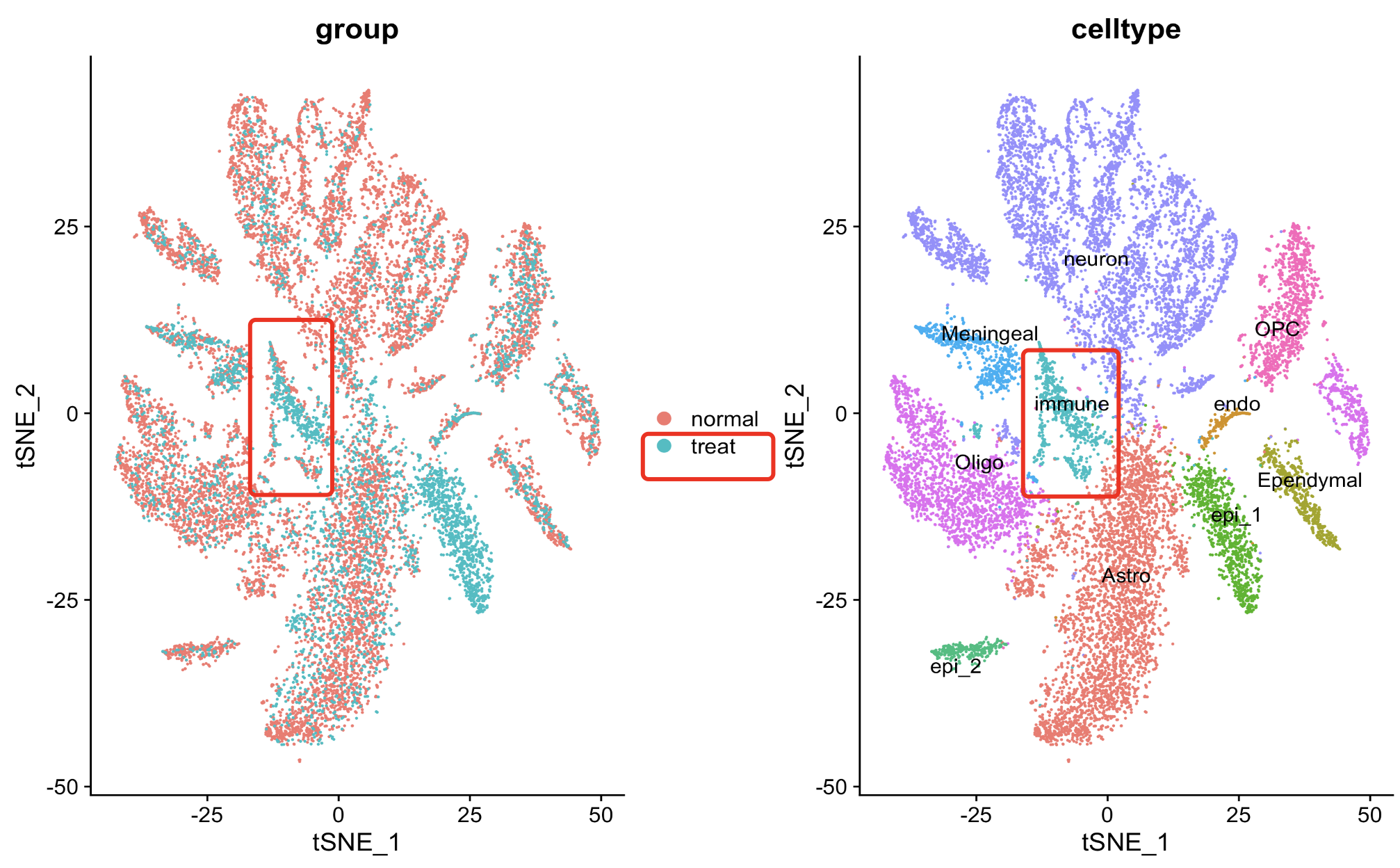
代码是:sce.all.int <- RunTSNE(sce.all.int, dims = 1:15, reduction = "harmony") table(sce.all.int$orig.ident) sce.all.int$group =ifelse( sce.all.int$orig.ident %in% 1:3,'normal','treat' ) table(sce.all.int$group ) DimPlot(sce.all.int, reduction = "tsne",raster = F, group.by = 'group', label = F ) + DimPlot(sce.all.int, reduction = "tsne",raster = F, group.by = 'celltype', label = T,repel = T)而且我们完完全全是使用了作者自己的特异性高表达的基因去定义生物学亚群,如下所示:
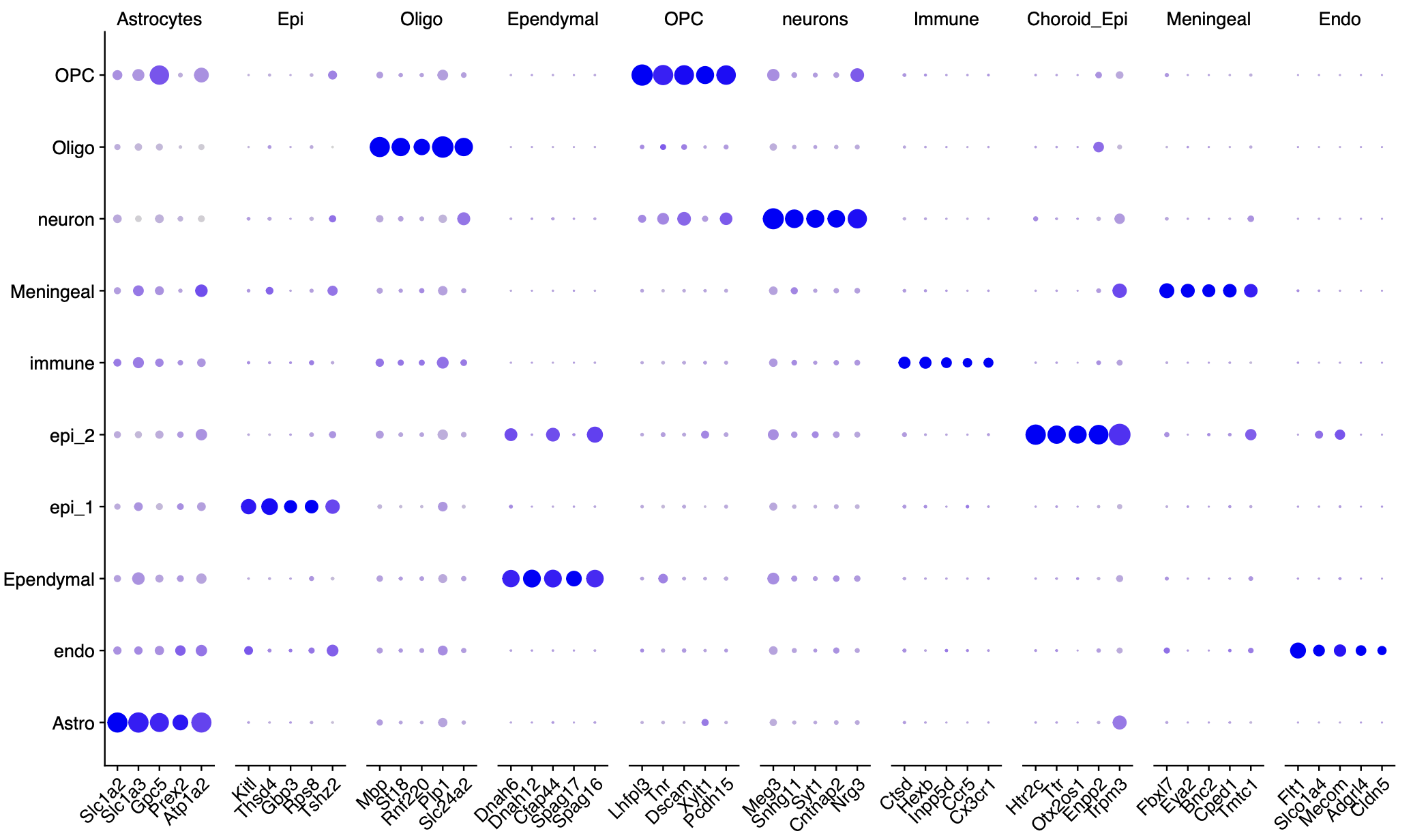
文献里面的基因列表组成的代码如下所示:Astrocytes = c("Slc1a2", "Slc1a3","Gpc5", "Prex2","Atp1a2") Epithelial = c("Kitl","Thsd4","Gbp3","Rps8","Tshz2") Oligodendrosytes = c("Mbp","St18","Rnf220","Plp1","Slc24a2") Ependymal = c( "Dnah6","Dnah12","Cfap44","Spag17","Spag16") Oligodendrosyte_progenitor = c("Lhfpl3","Tnr","Dscam","Xylt1","Pcdh15") Contaminating_neurons = c("Meg3","Snhg11","Syt1","Cntnap2","Nrg3") Immune_cells = c( "Ctsd","Hexb","Inpp5d","Ccr5","Cx3cr1") Choroid_plexus_epithelial_cells = c("Htr2c","Ttr","Otx2os1","Enpp2","Trpm3") Meningeal_cells=c("Fbxl7","Eya2","Bnc2","Cped1","Tmtc1") Endothelial =c("Flt1","Slco1a4","Mecom","Adgrl4","Cldn5") gene_list = list(Astrocytes,Epithelial,Oligodendrosytes,Ependymal,Oligodendrosyte_progenitor, Contaminating_neurons,Immune_cells,Choroid_plexus_epithelial_cells,Meningeal_cells,Endothelial) names(gene_list)=trimws( strsplit('Astrocytes,Epi,Oligo,Ependymal,OPC, neurons,Immune,Choroid_Epi,Meningeal,Endo',',')[[1]])所以也不存在命名冲突的情况,看起来是完美的复现了作者的结果, 但是如果仔细点看免疫细胞比例变化,如下所示:
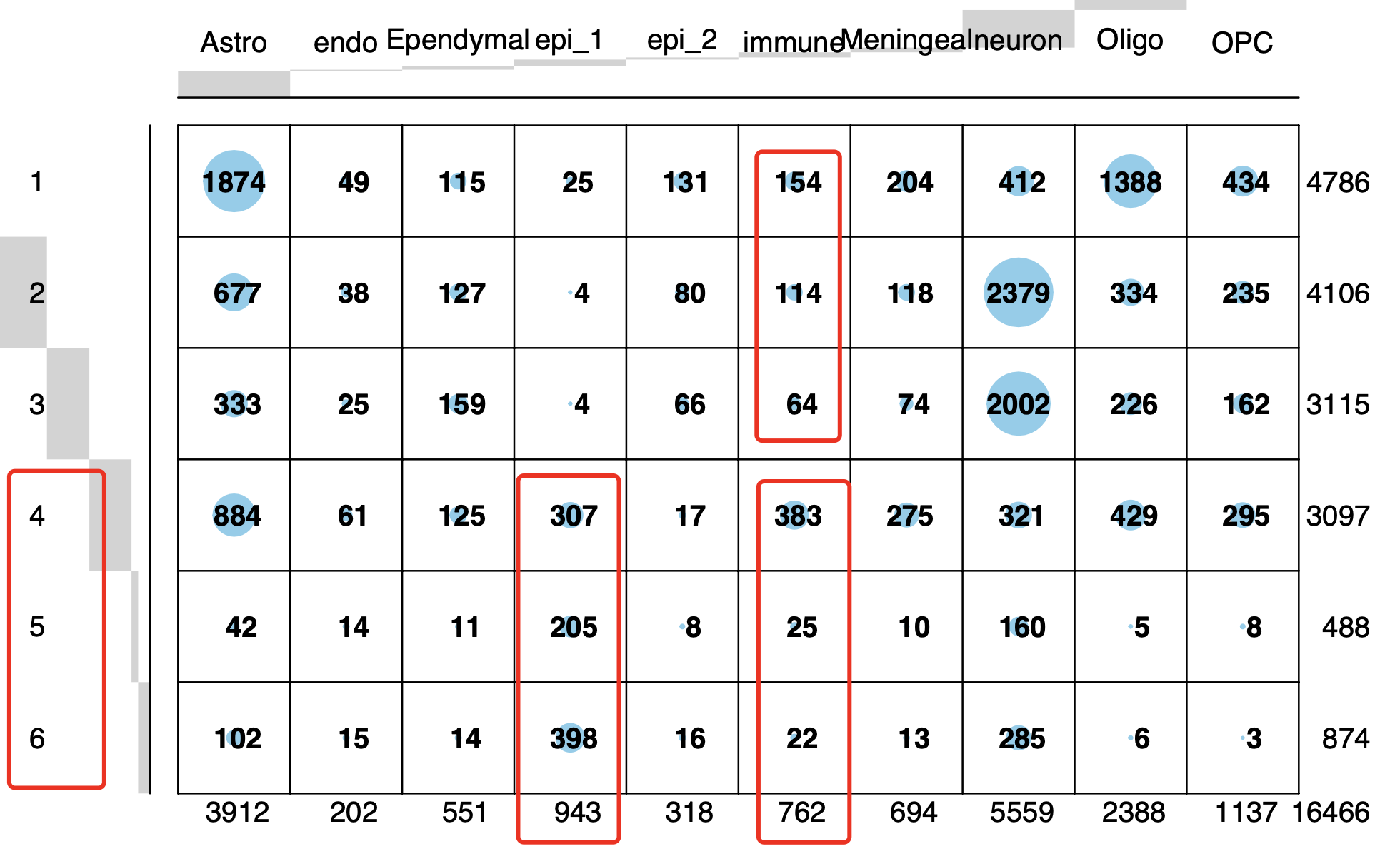
其实是有问题,确实是无论是从绝对数量和相对数量来说,在下面的3个样品组成的处理组里面都是远高于上面的3个样品的对照组,但是很明显的是它其实是由一个离群点样品造成的!那么我们该相信什么样的数据分析结果呢
这个时候无论是做什么统计学检验其实都有不够的,我这里相信了文章里面的脑转移造模成功的小鼠样品里面确实是有一群特异性的恶性的肿瘤上皮细胞,但是我没办法相信脑转移造模成功的小鼠样品里的免疫细胞就一定是有比例上升。
首先如果免疫细胞比例并没有可靠的上升,那么文章对免疫细胞细分的逻辑性就有点弱,然后如果是免疫细胞细分亚群就有问题, 那么文章最大的结论(肿瘤相关脑积水病发时,脉络丛肥大细胞增加)就站不住脚了。
求助广大网友
看看能不能读取这个GSE207546 数据集里面的单细胞核转录组是六个样品,进行降维聚类分群后,提取里面的免疫细胞亚群后继续细分,看看是不是脑转移造模成功的小鼠样品里的肥大细胞远高于正常组别。无论是从绝对数量还是相对数量,而且一定要注意是不是因为其中某个分组里面的一个样品导致的,如果是这样的离群点那么文章里面的结论就大打折扣啦!