通常情况下,如果大家提问说他们对自己的“照猫画虎”的生物信息学数据分析结果的准确性不确定的时候,我们会让他可视化后部分结果然后肉眼判断。因为为算法通常是给出来一个统计学指标,而我们高通量数据的分析通常是批量做大量的数据统计,会有大量的结果产生,这个其实就呼应了昨天的视频号直播: 生信分析和实验结果哪个更可靠(我和y叔的回答),y叔提到了因为存在大量的生物信息学初学者,他们对数据分析的认识不足导致数据分析出错误也会造成生信分析的不准确性。。。
比如,差异分析后一般来说有成百上千个上下调基因,如果对一个具体的基因是否有表达量差异这件事,而分析者都没办法确定,我们会让他做一个表达量箱线图。理论上肉眼看到的箱线图是金标准,如果你看到了某个基因表达量肉眼可见的降低,那么它就不应该是被算法判断为表达量升高!
不过,最近遇到了一个求助,是算法和肉眼冲突了,他查询自己的感兴趣基因的时候一直是可以看到箱线图里面的肉眼可见的两个分组的表达量差异,但是针对这个表达量矩阵进行算法层面的差异分析就始终发现它不是统计学显著的差异基因,甚至是相反的。
首先我们看看这个基因的具体的表达量数值,如下所示它是一个表达量下调基因:
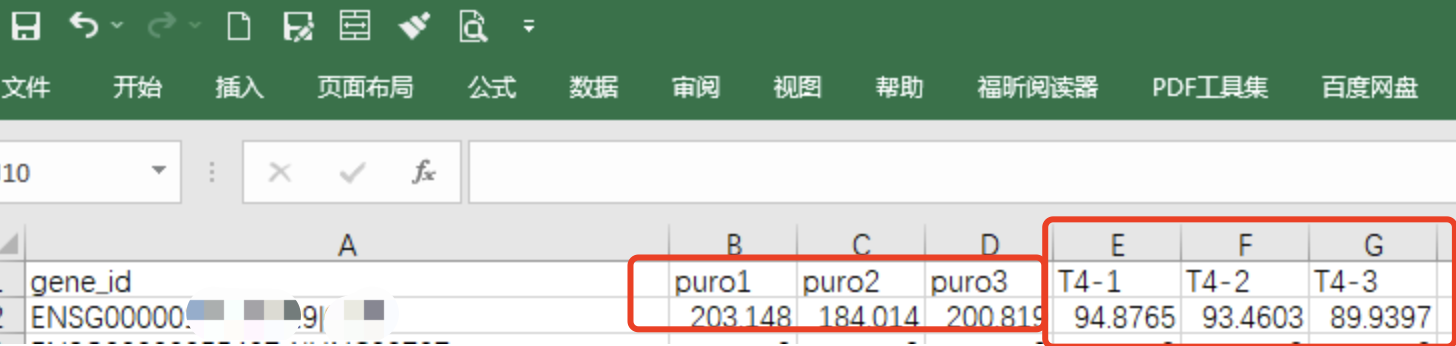
既然这个基因在两分组的TPM是下调很明显的,但是求助者说它用count做差异表达分析,却发现它被软件算法判定为显著上调。。。
起初,我是很不愿意相信的,就找对方拿到了原始的表达量矩阵文件,我亲自跑一下代码才发现居然真的是这样的!!!
# 魔幻操作,一键清空
rm(list = ls())
options(stringsAsFactors = F)
library(data.table)
mat <- read.table("inputs/all.id.txt",row.names = 1,
sep = "\t", header = T)
keep_feature <- rowSums (mat > 1) > 1
table(keep_feature)
ensembl_matrix <- mat[keep_feature, ]
library(AnnoProbe)
head(rownames(ensembl_matrix))
rownames(ensembl_matrix) = gsub('[.][0-9]*','',rownames(ensembl_matrix))
ids=annoGene(rownames(ensembl_matrix),'ENSEMBL','human')
ids=ids[!duplicated(ids$SYMBOL),]
ids=ids[!duplicated(ids$ENSEMBL),]
symbol_matrix= ensembl_matrix[match(ids$ENSEMBL,rownames(ensembl_matrix)),]
rownames(symbol_matrix) = ids$SYMBOL
group_list=ifelse(grepl('puro',colnames(symbol_matrix)),'control','case' )
group_list = factor(group_list,levels = c('control','case' ))
save(symbol_matrix,group_list,file = 'symbol_matrix.Rdata')
求助者给我的表达量矩阵是不公开的,所以我只能说截图给大家看看:
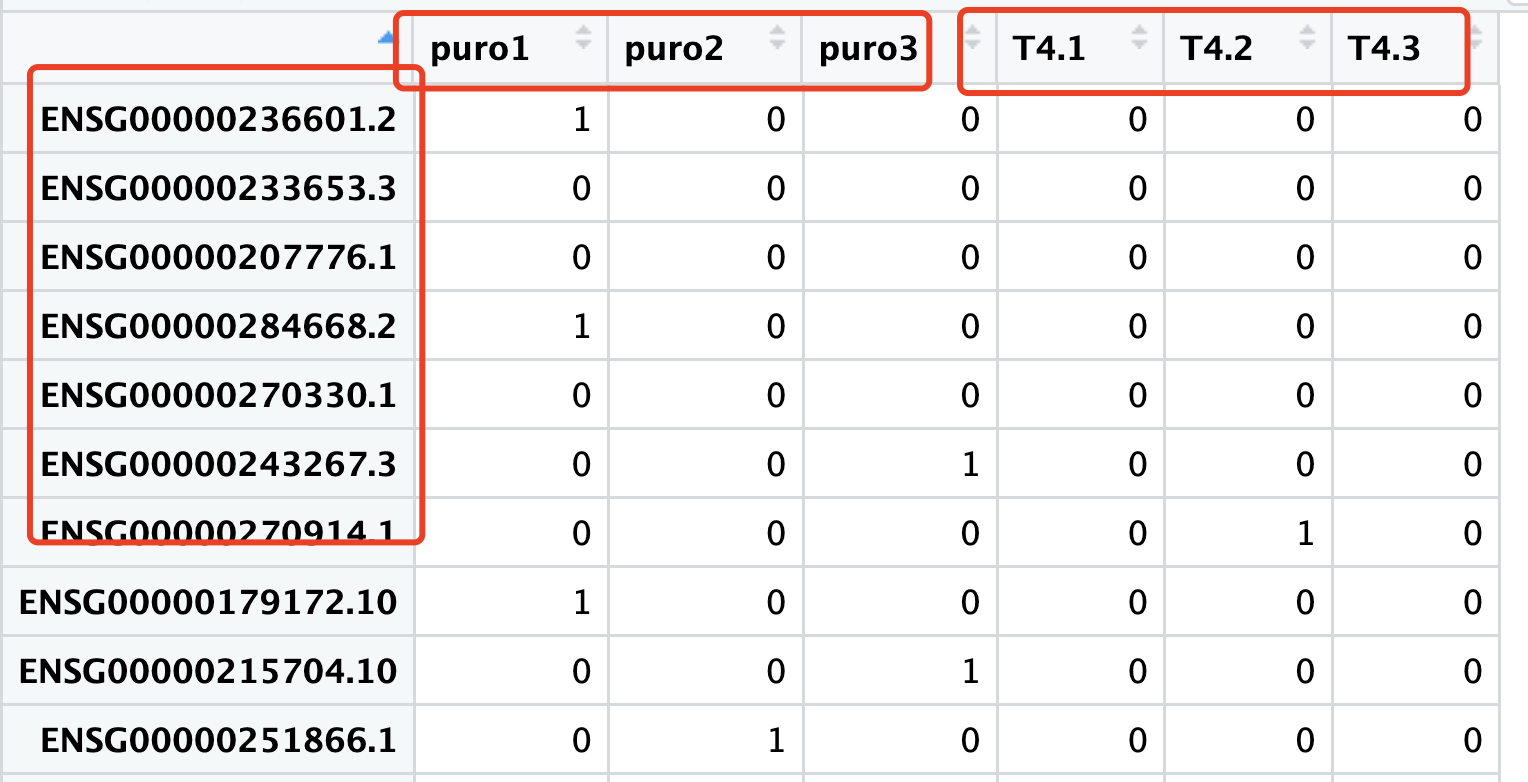
我也是简单的使用了 read.table 读取它:
read.table("inputs/all.id.txt",row.names = 1,
sep = "\t", header = T)
然后差异分析也是简单的DESeq2而已,如下所示:
library(DESeq2)
load(file = 'symbol_matrix.Rdata')
exprSet = symbol_matrix
(colData <- data.frame(row.names=colnames(exprSet),
group_list=group_list) )
dds <- DESeqDataSetFromMatrix(countData = exprSet,
colData = colData,
design = ~ group_list)
dds <- DESeq(dds)
res <- results(dds,
contrast=c("group_list",
levels(group_list)[2],
levels(group_list)[1]))
resOrdered <- res[order(res$padj),]
head(resOrdered)
DEG =as.data.frame(resOrdered)
DEG_deseq2 = na.omit(DEG)
save(DEG_deseq2,
file = 'DEG_deseq2.Rdata' )
我们简单的解析DESeq2的差异分析结果,可以看到箱线图里面的这个基因是表达量下调,但是它的log2FoldChange是大于0 的,这个 log2FoldChange=0.8 相当于是说这个基因在case组的表达量是control组的 1.74倍啦,妥妥的上调。。。
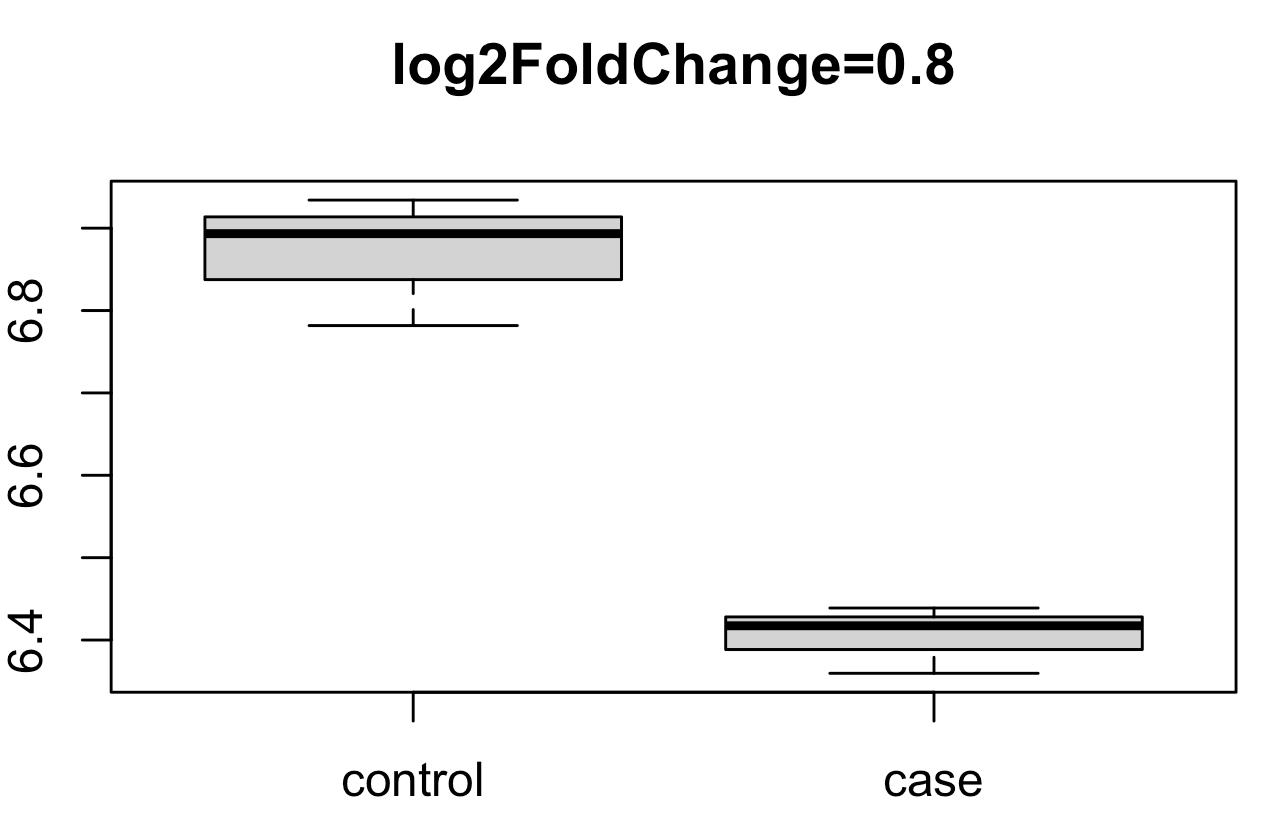
那么,这个冲突是为什么呢?log2FoldChange理论上是非常容易计算获取啦:
dat = log2(edgeR::cpm(symbol_matrix)+1)
dat[1:4,1:4]
DEG=DEG_deseq2[rownames(dat),]
DEG$df=rowMeans(dat[,4:6]) - rowMeans(dat[,1:3])
plot(DEG$df,DEG$log2FoldChange)
df = DEG[DEG$padj < 0.01,]
library(ggpubr)
ggscatter(df, x = "df", y = "log2FoldChange",
color = "black", shape = 21, size = 3, # Points color, shape and size
add = "reg.line", # Add regressin line
add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
conf.int = TRUE, # Add confidence interval
cor.coef = TRUE, # Add correlation coefficient. see ?stat_cor
cor.coeff.args = list(method = "pearson", label.sep = "\n")
)
可以看到DESeq2的差异分析结果里面的log2FoldChange并不是简简单单case组的表达量的平均值去跟control组对比,而是经过了一定程度的矫正 ,
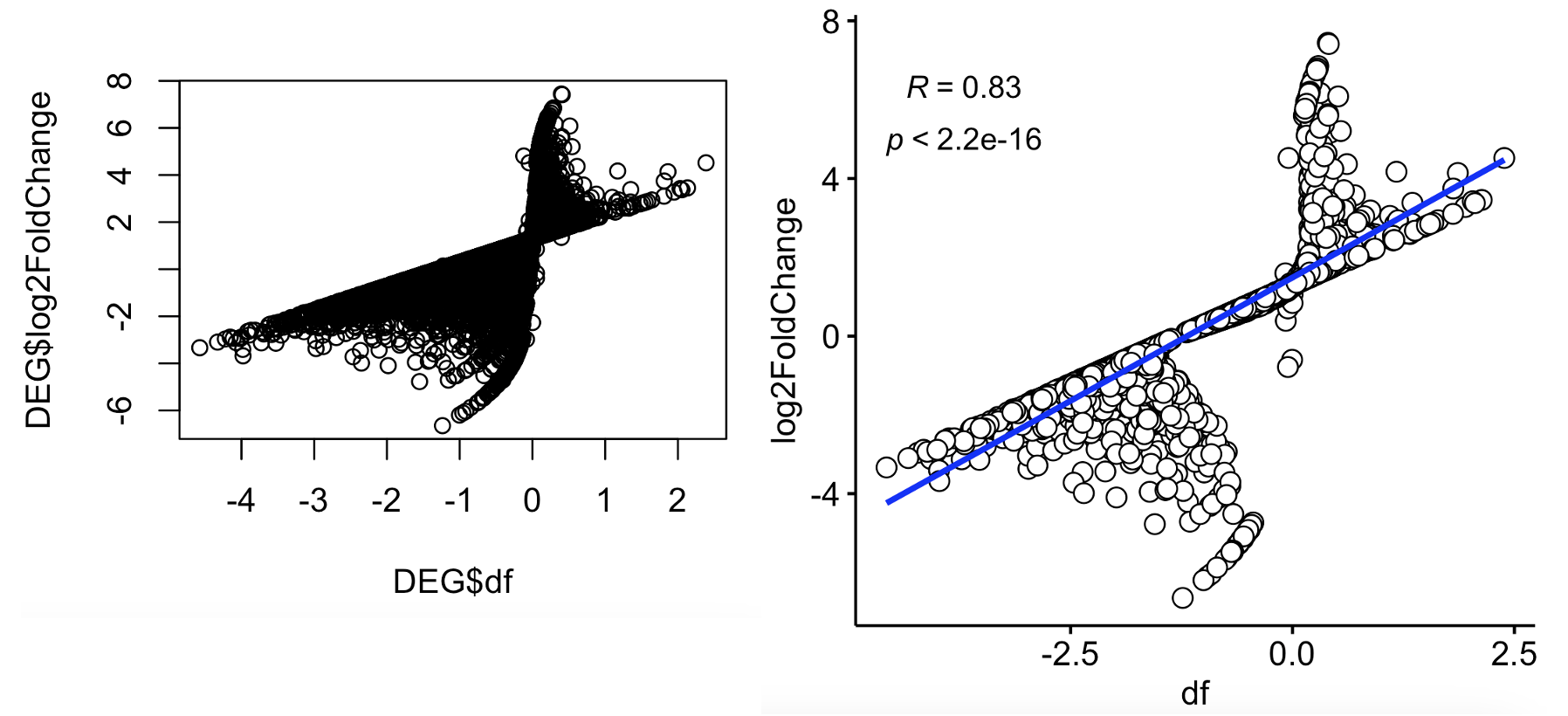
确实是会出现一些基因在x和y轴出现了正负值的反转,比如下面的放大的:
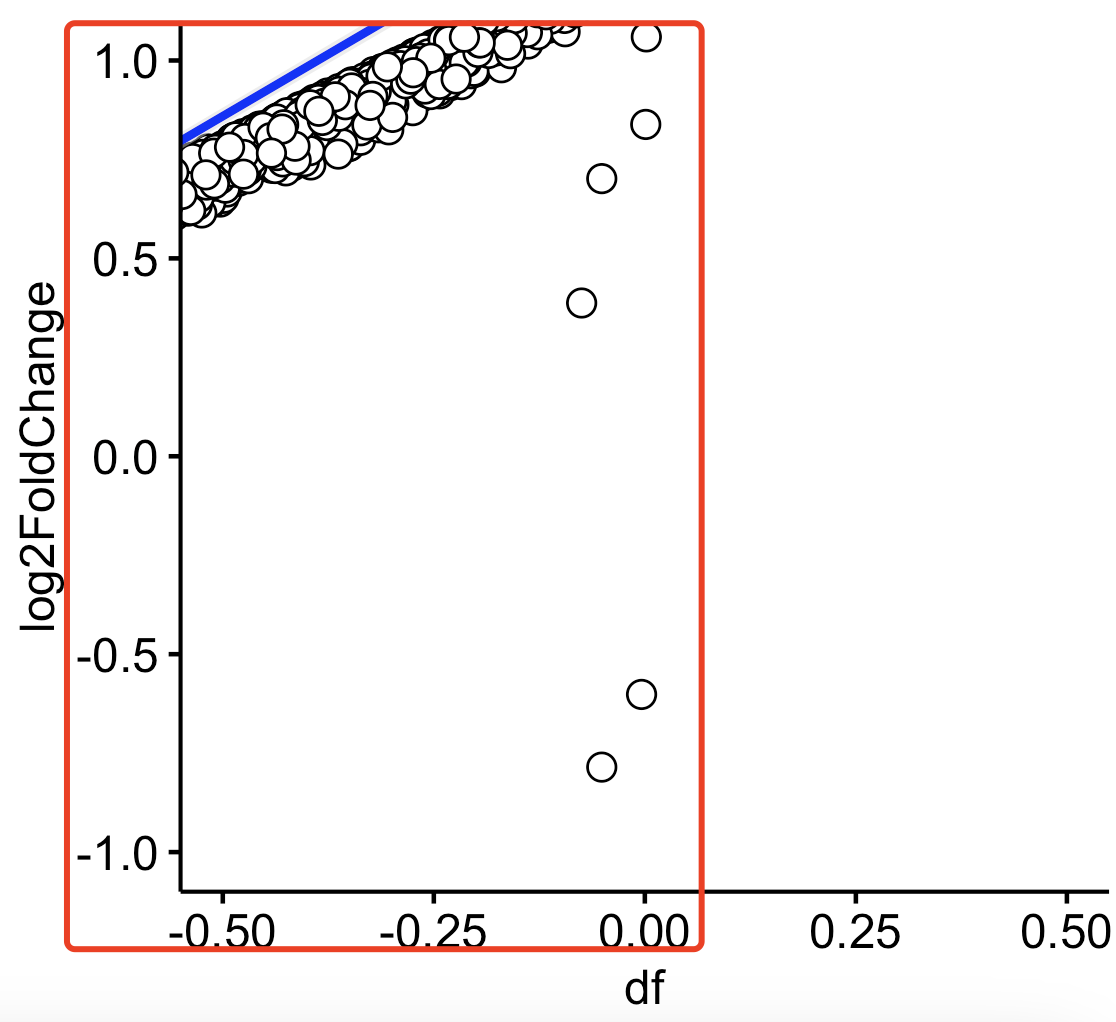
这样的基因数量还不少:
> table(df$log2FoldChange <0,df$df<0)
FALSE TRUE
FALSE 1040 2475
TRUE 0 3892
如果想彻底理解当算法和肉眼冲突了肿么办,就需要熟读这两个文献:
- https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-14-91
- https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02648-4
其实转录组测序的数据分析已经是非常标准化啦
我们给大家的示例流程也是主流CNS文章采用的,hisat2比对后接着是featureCounts定量,然后是DESeq2的差异分析,比如文章:《Caloric Restriction Reprograms the Single-Cell Transcriptional Landscape of Rattus Norvegicus Aging》,就是如此:

因为太常规了,所以都是明码标价的,转录组产品线