大家最常接触的转录组数据分析教学环节都是二分组,处理和对照,疾病和正常,这样的差异分析很容易理解。但是真实的科研往往是更复杂一点,前面我们分享了:时间序列转录组多次差异分析以及时序分析,是不同时间点处理的肿瘤细胞系表达量芯片数据。
然后我们把这个代码移植到了转录组测序数据集,详见:表达量芯片的代码当然是可以移植到转录组测序数据分析,它实际上并不是真正的时间序列采样的转录组,仅仅是因为疾病的状态具有连续性而已。以看到:
> as.data.frame(table(phe$`group in paper:ch1`))
Var1 Freq
1 control 10
2 NAFL 51
3 NASH_F0-F1 34
4 NASH_F2 53
5 NASH_F3 54
6 NASH_F4 14
分组需要结合背景知识,这个 Fibrosis-4指数(FIB-4) 用于评估NAFLD患者的肝纤维化程度,而不是NASH本身的严重程度。它使用年龄、AST(天门冬氨酸转氨酶)和ALT(丙氨酸转氨酶)水平以及血小板计数来计算。FIB-4指数通常用于筛查哪些NAFLD患者可能有肝纤维化,而不是评估NASH的严重程度。
但是今天的单细胞天地公众号分享了一个单细胞数据集(GSE168113),就是完美的病毒感染与否的时间序列采样的转录组,详见:来源于多个物种的单细胞转录组表达量矩阵如何处理,虽然说它是单细胞层面的表达量矩阵,但是我们很容易做出来pseudobulk矩阵,使用rowSums方法即可,这个是非常容易理解的,我在之前分享了:单细胞层面的表达量差异分析到底如何做。
但是这个文章对单细胞数据集(GSE168113)的pseudobulk矩阵仅仅是做了一个PCA分析,说明他们的病毒感染与否的分组的差异是大于时间序列差异而已。
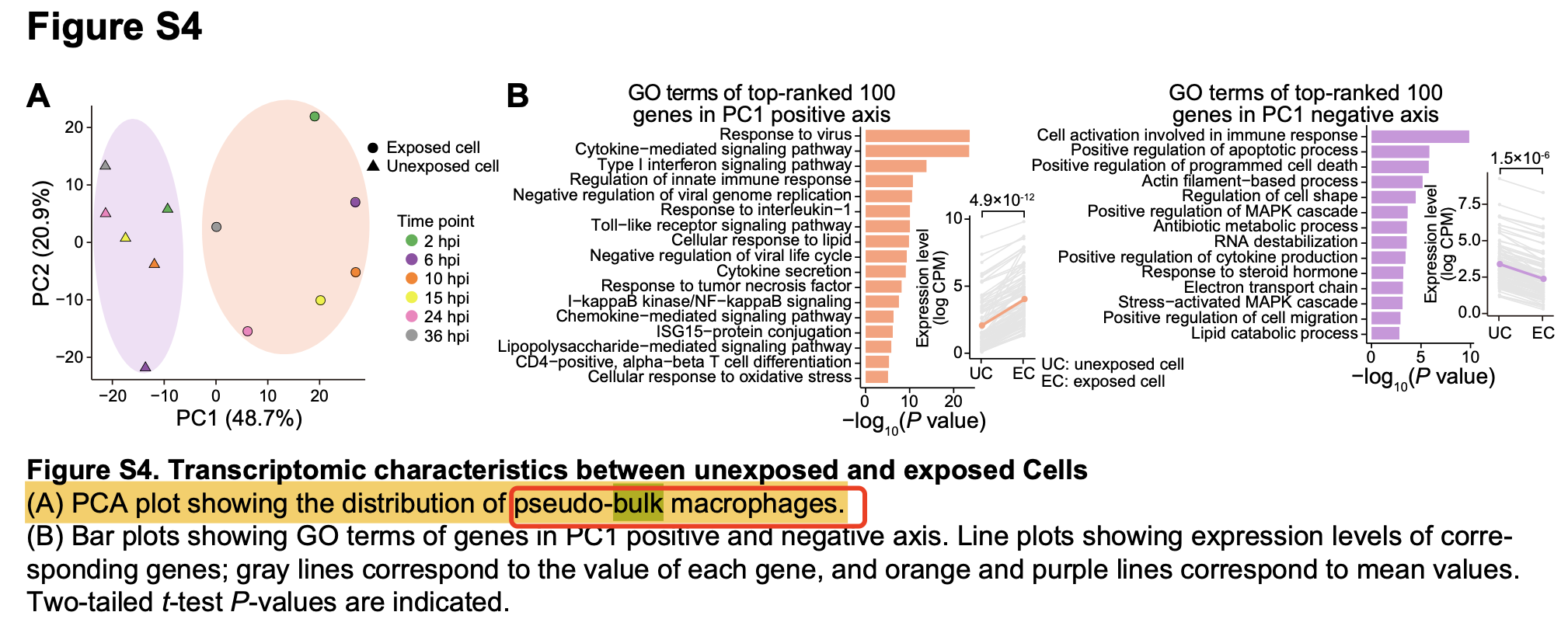
感兴趣的可以去阅读文章《Transcriptome profiling in swine macrophages infected with African swine fever virus at single-cell resolution》,而且我把它接下来常规的降维聚类分群详见:来源于多个物种的单细胞转录组表达量矩阵如何处理,现在把表达量矩阵给大家,因为分组信息,时间序列信息,非常清晰,很适合做前面的mfuzz代码处理,详见:表达量芯片的代码当然是可以移植到转录组测序数据分析。
GSM5129158 UninfectedGroup_T0
GSM5129159 UninfectedGroup_T2
GSM5129160 UninfectedGroup_T6
GSM5129161 UninfectedGroup_T10
GSM5129162 UninfectedGroup_T15
GSM5129163 UninfectedGroup_T24
GSM5129164 UninfectedGroup_T36
GSM5129165 InfectedGroup_T2
GSM5129166 InfectedGroup_T6
GSM5129167 InfectedGroup_T10
GSM5129168 InfectedGroup_T15
GSM5129169 InfectedGroup_T24
GSM5129170 InfectedGroup_T36
我这里制作pseudobulk矩阵的代码是 :
table(sce.all.int$celltype)
sce.all=sce.all.int[,sce.all.int$celltype=='mac']
av <-AverageExpression(sce.all ,
group.by = 'orig.ident',
assays = "RNA")
av=av[[1]]
dim(av)
av[1:4,1:4]
cg=names(tail(sort(apply(av, 1, sd)),1000))
pheatmap::pheatmap(cor(av[cg,]))
ct = do.call(
cbind,lapply(unique(sce.all$orig.ident), function(x){
#x = unique(sce.all$orig.ident)[1]
kp = sce.all$orig.ident %in% x
print(table(kp))
rowSums( as.matrix( sce.all@assays$RNA@counts[, kp] ))
})
)
colnames(ct)=unique(sce.all$orig.ident)
dim(ct)
ct[1:4,1:4]
save(av,ct,file = 'input_for_deg.Rdata')
学徒作业
大家首先拿到了我上面给大家制作好的 input_for_deg.Rdata 文件后,进行简单的质量控制,看看是不是跟文章那样的病毒感染分组的差异是大于时间序列的,然后进行各种各样的组合差异分析。
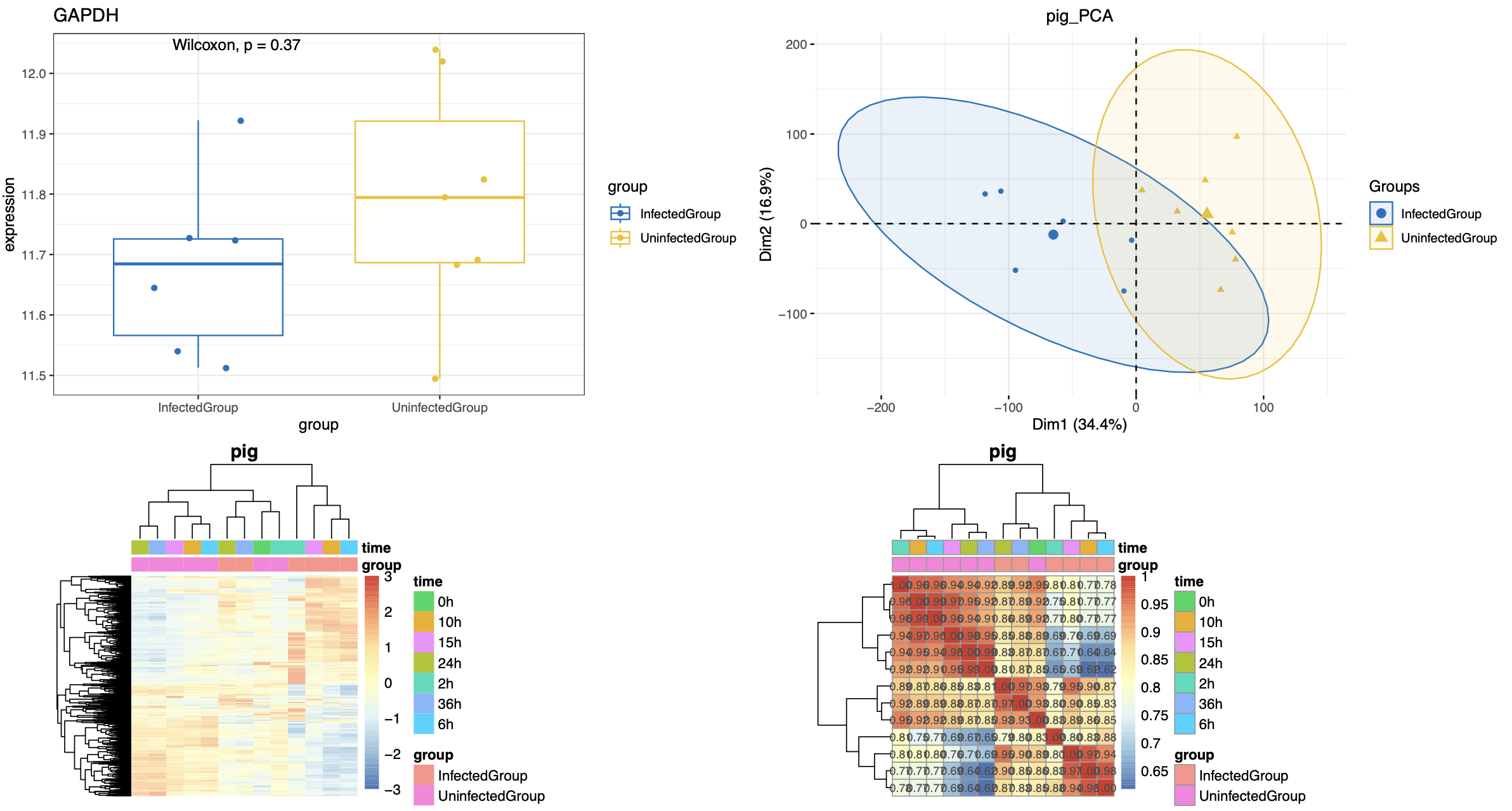
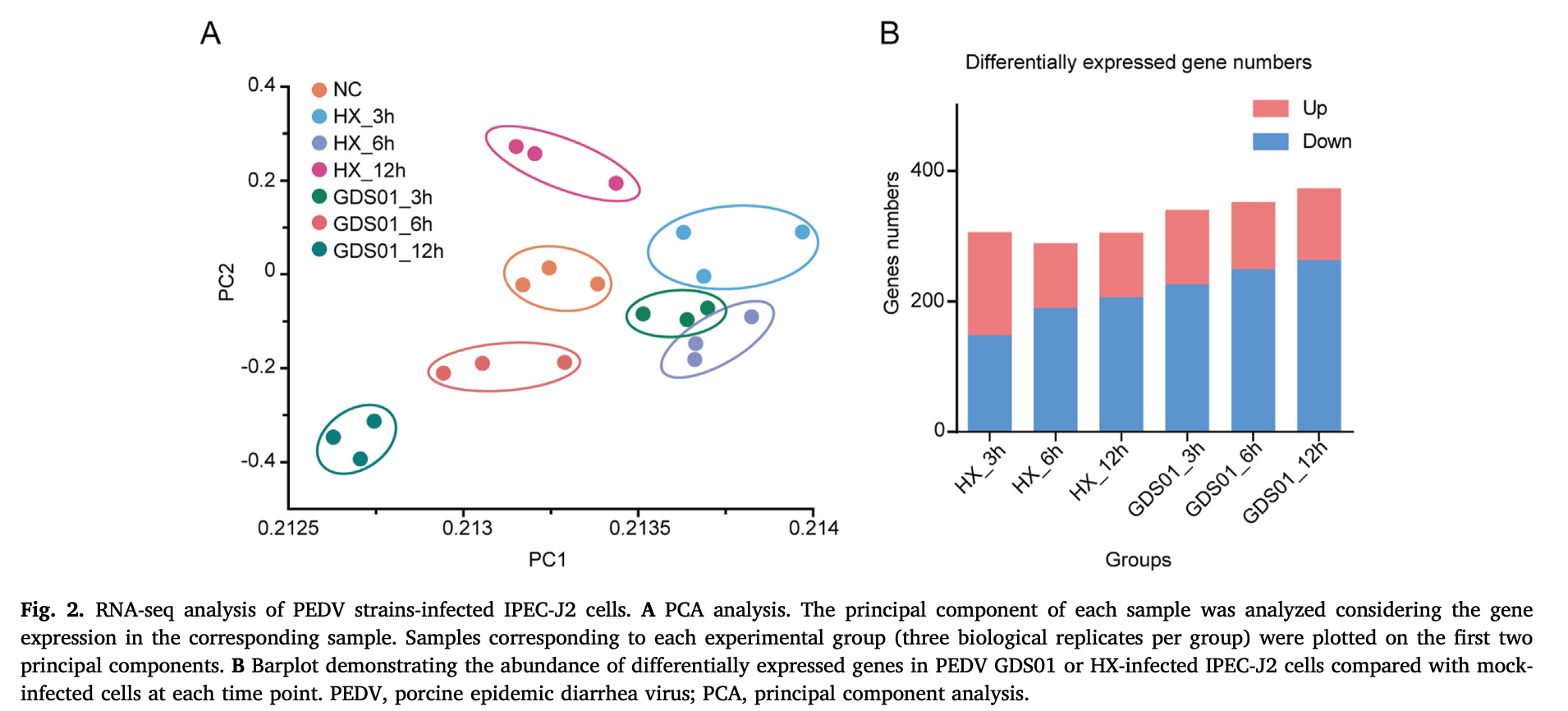
然后就可以参考2022的文章:《Genome-wide transcriptome analysis of porcine epidemic diarrhea virus virulent or avirulent strain-infected porcine small intestinal epithelial cells》
各种各样的差异分析并且汇总
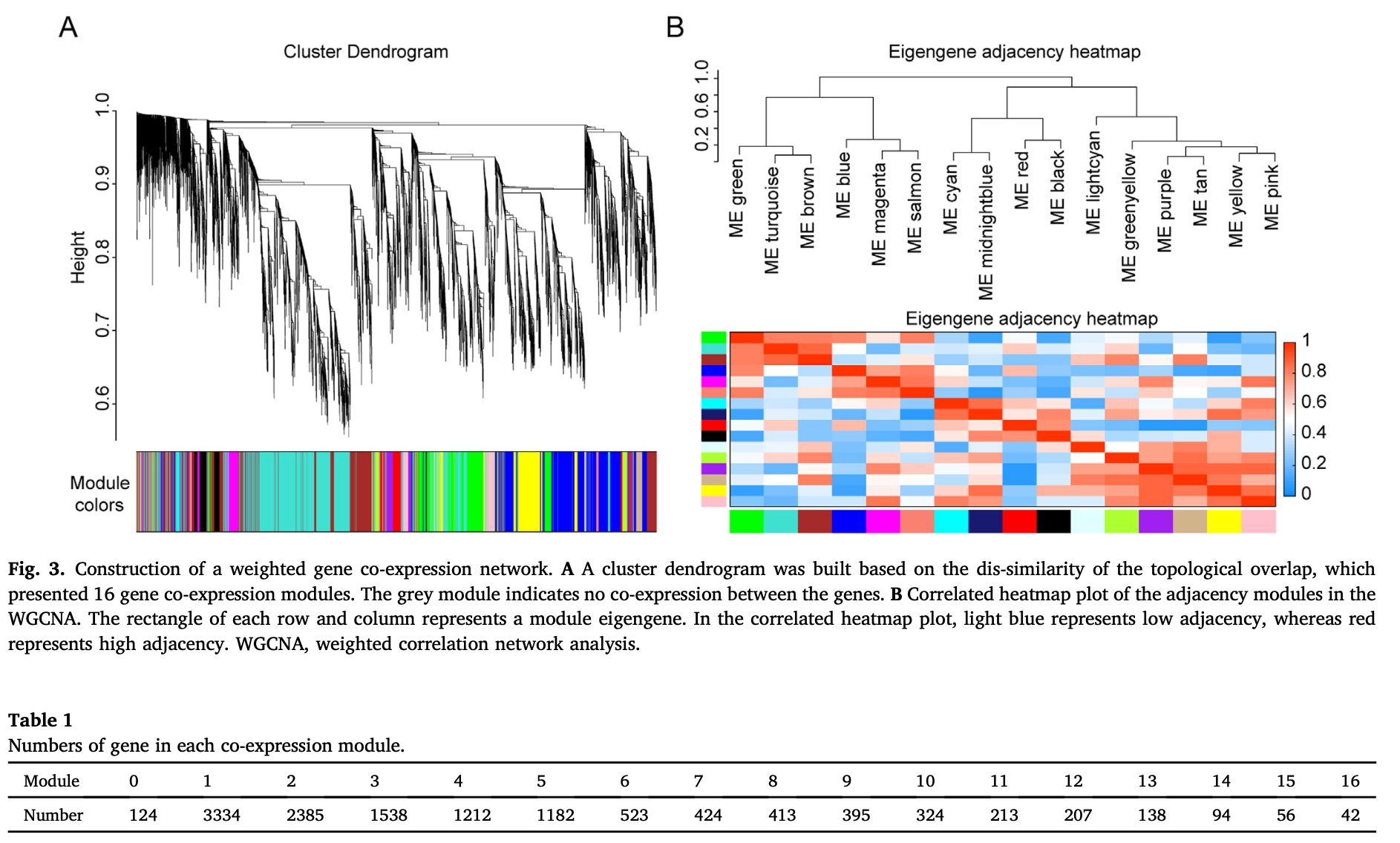
wgcna分析
mfuzz分析
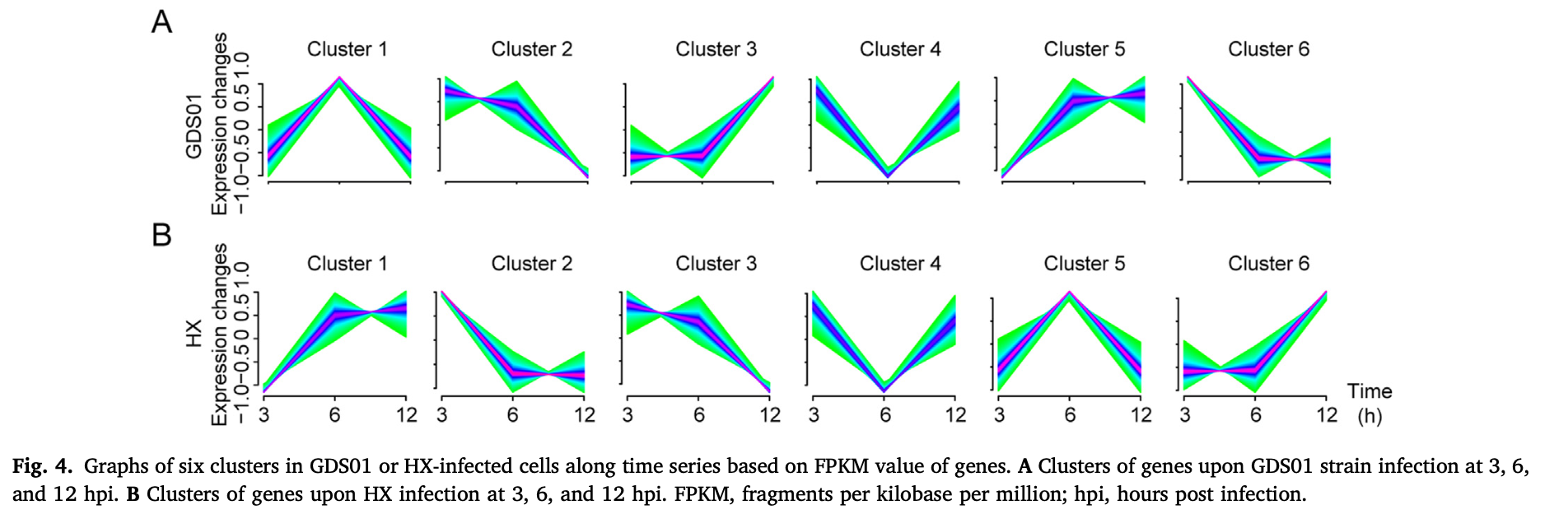
上面的mfuzz分析和wgcna分析,都是可以把基因划分成为了不同的分组,每个分组就是一个基因集合,基因集合就可以进行go和kegg这样的功能数据库注释。