做过单细胞转录组数据分析的小伙伴都知道,多分组多样品的单细胞项目很容易就十几万个单细胞了所以第一层次降维聚类分群就有十个左右亚群,然后每个亚群又可以细分十几个,算起来就是近百的单细胞亚群。
这样的话图表会非常多,但是数据分析的终极目标是发文章,而文章就必须要有故事性有落脚点。接下来我们就系统性梳理一下使用生存分析来辅助判定关键单细胞亚群。这里我们采用的仍然是2022的肺腺癌相关的文章,标题是:《Delineating the dynamic evolution from preneoplasia to invasive lung adenocarcinoma by integrating single-cell RNA sequencing and spatial transcriptomics》,肺癌单细胞数据集也有好几十个了,拿到表达量矩阵后的第一层次降维聚类分群通常是:
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibo or CD31+,PECAM1,endo)
参考我前面介绍过 CNS图表复现08—肿瘤单细胞数据第一次分群通用规则,这3大单细胞亚群构成了肿瘤免疫微环境的复杂。其实这个文章的第一层次降维聚类分群详见:换一个分析策略会导致文章的全部论点都得推倒重来吗,然后文章针对上皮细胞进行细分后,使用inferCNV判断了恶性的亚群,接下来就使用生存分析从恶性亚群里面的挑选了增殖亚群(UBE2C+),因为它这个亚群的基因可以在tcga数据库的luad数据集里面的有统计学显著的生存分析意义。如下所示:
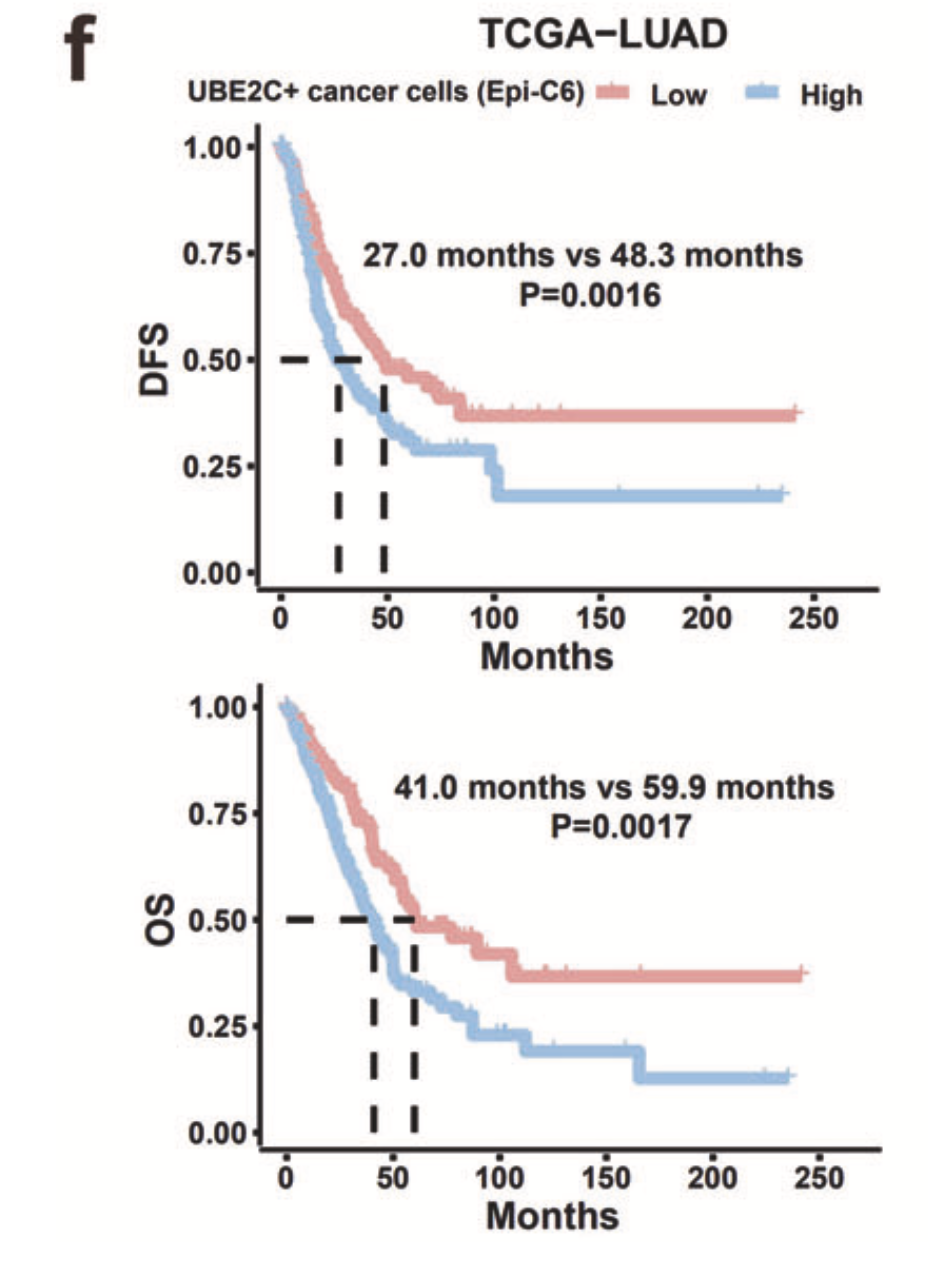
生存分析三步走
生存分析主要是需要一个病人队列而且病人的生存时间信息以及其最后的状态信息是齐全的,然后我们需要病人的表达量矩阵信息,这样的话就可以从矩阵里面挑选感兴趣的基因或者说基因列表去矩阵里面打分后,把病人高低分组后做生存分析来判断其重要性。
这里我们采用ucsc的xena浏览器下载tcga数据库的luad数据集表达量矩阵,每个癌症都有固定的数据文件格式:
比如TCGA-LAML,如下所示:
https://gdc-hub.s3.us-east-1.amazonaws.com/download/TCGA-LAML.htseq_counts.tsv.gz
https://gdc-hub.s3.us-east-1.amazonaws.com/download/TCGA-LAML.GDC_phenotype.tsv.gz
https://gdc-hub.s3.us-east-1.amazonaws.com/download/TCGA-LAML.survival.tsv
https://gdc-hub.s3.us-east-1.amazonaws.com/download/TCGA-LAML.mutect2_snv.tsv.gz
很容易批量下载,也很容易根据感兴趣的癌症来下载对应的文件。
第1步:格式化表达量矩阵文件
下面的代码很容易理解,需要自己熟悉这个TCGA-LUAD.htseq_counts.tsv.gz文件的规则即可:
rm(list=ls())
library(data.table)
dat <- data.table::fread('TCGA-LUAD.htseq_counts.tsv.gz',
data.table = F)
head(dat[,1:4])
tail(dat[,1:4])
dat = dat[1:(nrow(dat)-5),]
rownames(dat) = dat$Ensembl_ID
a = dat
a = a[,-1]
##逆转 log
a = as.matrix(2^a - 1)
# 用apply转换为整数矩阵
head(a[,1:4])
tail(a[,1:4])
colSums(a)/1e6
# 普通转录组定量 20m
# 想看融合基因,可变剪切,100M
exp = apply(a, 2, as.integer)
rownames(exp) = rownames(dat)
exp= log(edgeR::cpm(exp)+1)
library(stringr)
head(rownames(exp))
library(AnnoProbe)
library(tinyarray)
rownames(exp) = substr(rownames(exp), 1, 15)
re = annoGene(rownames(exp),ID_type = "ENSEMBL");head(re)
exp = trans_array(exp,ids = re,from = "ENSEMBL",to = "SYMBOL")
head(exp[,1:4])
tail(exp[,1:4])
proj='tcga-luad'
save(exp,file = paste0(proj,".htseq_counts.rdata") )
第2步:整理临床信息
做生存分析的时候,其实只需要表达量矩阵里面的肿瘤样品即可,所以简单的过滤一下。然后临床信息里面我们也是简单的取 OS 即可:
rm(list=ls())
proj='tcga-luad'
load(file = paste0(proj,".htseq_counts.rdata") )
Group = ifelse(as.numeric(str_sub(colnames(exp),14,15)) < 10,'tumor','normal')
Group = factor(Group,levels = c("normal","tumor"))
print(table(Group))
# 生存分析只需要tumor样品即可
exprSet = exp[,Group=='tumor']
clinical = read.delim('TCGA-LUAD.GDC_phenotype.tsv.gz',
fill = T,header = T,sep = "\t")
surv = read.delim('TCGA-LUAD.survival.tsv',header = T)
library(tidyverse)
meta = left_join(surv,clinical,by = c("sample"= "submitter_id.samples"))
head(meta[,1:4])
tail(meta[,1:4])
print(dim(meta))
#去掉生存信息不全或者生存时间小于30天的样本,样本纳排标准不唯一,且差别很大.
k1 = meta$OS.time >= 30
k2 = !(is.na(meta$OS.time)|is.na(meta$OS))
meta = meta[k1&k2,]
meta = meta[,c(
'sample',
'OS',
'OS.time'
)]
colnames(meta)=c('ID','event','time')
meta$time = meta$time/30
rownames(meta) <- meta$ID
s = intersect(rownames(meta),colnames(exprSet))
exprSet = exprSet[,s]
meta = meta[s,]
identical(rownames(meta),colnames(exprSet))
save(exprSet,meta,file = paste0(proj,".for_survival.rdata") )
前面的表达量矩阵跟临床生存分析已经是对应好了,就可以进行后续的生存分析啦
第3步:批量cox分析
这个代码也是七年前的了,蛮简单的的一个循环而已
rm(list=ls())
library(survival)
library(survminer)
library(ggstatsplot)
proj='tcga-luad'
load(file = paste0(proj,".for_survival.rdata") )
# 1. prepare data for coxph----
## 批量生存分析使用coxph回归方法
exprSet[1:4,1:4]
phe=meta
head(phe)
mySurv <- with(phe, Surv(time, event))
survival_dat=phe
cox_results <-apply(exprSet , 1 , function(gene){
# gene= as.numeric(exprSet[1,])
group=ifelse(gene>median(gene),'high','low')
group=factor( group,levels = c('low','high'))
if( length(table(group))<2)
return(NULL)
survival_dat <- data.frame(group=group,# stage=phe$stage,
stringsAsFactors = F)
m=coxph(mySurv ~ group,
# mySurv ~ stage+ group, # 如果有交叉变量
data = survival_dat)
beta <- coef(m)
se <- sqrt(diag(vcov(m)))
HR <- exp(beta)
HRse <- HR * se
#summary(m)
tmp <- round(cbind(coef = beta, se = se, z = beta/se,
p = 1 - pchisq((beta/se)^2, 1),
HR = HR, HRse = HRse,
HRz = (HR - 1) / HRse, HRp = 1 - pchisq(((HR - 1)/HRse)^2, 1),
HRCILL = exp(beta - qnorm(.975, 0, 1) * se),
HRCIUL = exp(beta + qnorm(.975, 0, 1) * se)), 3)
return(tmp['grouphigh',])
})
# 2. specify the value----
cox_results=t(cox_results)
head(cox_results)
cox_results=cox_results[order(cox_results[,'HR'],decreasing = T),]
table(cox_results[,4]<0.01)
table(cox_results[,4]<0.05)
save(cox_results,
file = 'batch_cox_results.Rdata')
load( file = 'batch_cox_results.Rdata')
值得注意的是结果矩阵里面很多基因的HR值非常离谱,其实是因为这个基因在样品队列里面的表达量失衡,比如它仅仅是在少量的几个预后非常差的病人里面有表达量在所有的其它病人里面都是0值,这样的生存分析结果大概率是不可靠的!
基于单细胞转录组的生存分析
第1步:根据单细胞亚群基因集在肿瘤病人表达量矩阵里面进行gsva打分
如果是想看单个基因是否有统计学显著的生存分析意义其实前面的批量cox分析就可以提取结果了,但是我们的单细胞亚群基因集结果里面的基本上每个亚群都是几十个高表达量特异性基因,详见:换一个分析策略会导致文章的全部论点都得推倒重来吗,就知道如何输出如下所示的cosg_celltype__marker_cosg.Rdata文件啦:
rm(list=ls())
library(survival)
library(survminer)
library(ggstatsplot)
library(gplots)
library(ggplot2)
library(pheatmap)
library(clusterProfiler)
library(org.Hs.eg.db)
library(GSVA)
library(GSEABase)
# 1. 载入表达量矩阵和临床信息 ----
proj='tcga-luad'
load(file = paste0(proj,".for_survival.rdata") )
exprSet[1:4,1:4]
phe=meta
head(phe)
mySurv <- with(phe, Surv(time, event))
survival_dat=phe
# 2. creat geneset----
# load('../../paper-figures/cosg_celltype__marker_cosg.Rdata')
load('cosg_celltype__marker_cosg.Rdata')
head(marker_cosg$names)
deg_list = as.list(marker_cosg$names)
names(deg_list)
deg_list
gs = lapply(deg_list, toupper)
geneset <- GeneSetCollection(mapply(function(geneIds, keggId) {
GeneSet(geneIds, geneIdType=EntrezIdentifier(),
collectionType=KEGGCollection(keggId),
setName=keggId)
}, gs, names(gs)))
geneset
# 3. run gsva----
X=as.matrix(exprSet)
es.max <- gsva(X, geneset,
mx.diff=FALSE, verbose=FALSE,
parallel.sz=4)
es.max[1:4, 1:4]
pheatmap(es.max)
第2步:根据gsva打分值高低分组进行生存分析
一般来说, 就是根据gsva打分的中位值高低分组进行生存分析,代码如下所示:
# 4. 根据gsva结果高低分组后批量生存分析 ----
es.max[1:4, 1:4]
splots <- list()
g = 1
for (i in names(deg_list) ) {
# i = names(deg_list) [1]
subset = paste0('cluster_',i)
print(subset)
v = as.numeric(es.max[i,]) #每一个亚群表达量。
sub_group <- ifelse( v < 0,"low","high") #如果表达量小于0的话,就定义为low。gsva处理过表达量。0.几左右
table(sub_group)
phe$sub_group=sub_group
# Fit survival curves
require("survival")
fit <- survfit(Surv(time, event) ~ sub_group, data = phe)
library("survminer")
survp <- ggsurvplot(fit, data = phe,
surv.median.line = "hv", # Add medians survival
pval = TRUE, # Add p-value and tervals
conf.int = TRUE, # Add the 95% confidence band
risk.table = TRUE, # Add risk table
tables.height = 0.2,
tables.theme = theme_cleantable(),
palette = "jco",
ggtheme = theme_bw(),
title = subset)
print(survp)
splots[[g]] <- survp
g = g + 1
}
length(splots)
x1 = ceiling(sqrt(length(splots)))
y1 = x1
all_plot <- arrange_ggsurvplots(splots,
print = F,
ncol = x1, nrow = y1,
risk.table.height = 0.3,
surv.plot.height = 0.7)
# all_plot
x2=5*x1
y2=5*y1
prefix=''
pro=''
ggsave(all_plot, #path = prefix,
filename = paste0(pro, 'all_survival_plot.pdf'),
width = x2,height = y2)
就可以看到如下所示的每个单细胞亚群的生存分析结果,很明显跟文章类似的,也是增殖亚群(UBE2C+) 它这个亚群的基因可以在tcga数据库的luad数据集里面的有统计学显著的生存分析意义。
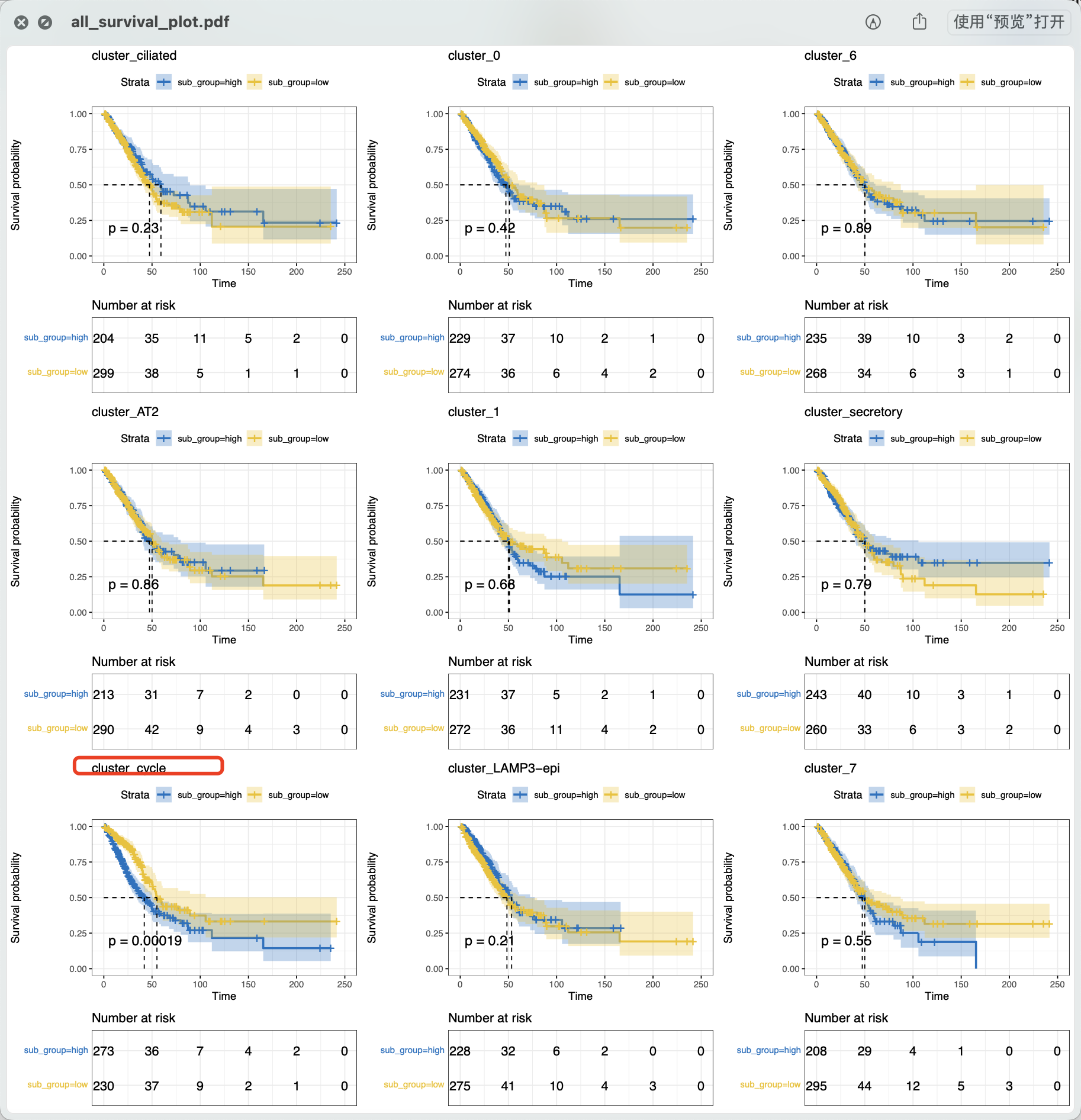
第3步:根据gsva打分值进行取巧分组进行生存分析
如果是上面的根据gsva打分的中位值高低分组进行生存分析都没有生存分析统计学显著意义,但是又想看看每个亚群的具体的到底是保护因子还是风险因子,也可以使用surv_cutpoint函数哦:
## cut point
## cutpoint
head(phe)
csplots <- list()
cg = 1
for (i in names(deg_list) ) {
# i = names(deg_list) [1]
subset = paste0('cluster_',i)
print(subset)
v = as.numeric(es.max[i,]) #每一个亚群表达量。
phe$v <- v
head(phe)
sur.cut <- surv_cutpoint(phe,
time= 'time',
event = 'event' ,
variables = 'v' )
sur.cat <- surv_categorize(sur.cut)
head(sur.cat)
sfit <- survfit(Surv(time, event) ~ v, data = sur.cat)
p_surv_cut <- ggsurvplot(sfit, data = phe,
surv.median.line = "hv", # Add medians survival
pval = TRUE, # Add p-value and tervals
conf.int = TRUE, # Add the 95% confidence band
risk.table = TRUE, # Add risk table
tables.height = 0.2,
tables.theme = theme_cleantable(),
palette = "jco",
ggtheme = theme_bw(),
title = subset)
print(p_surv_cut)
csplots[[cg]] <- p_surv_cut
cg = cg + 1
}
length(csplots)
x1 = ceiling(sqrt(length(csplots)))
y1 = x1
all_plot <- arrange_ggsurvplots(csplots,
print = F,
ncol = x1, nrow = y1,
risk.table.height = 0.3,
surv.plot.height = 0.7)
# all_plot
x2=5*x1
y2=5*y1
ggsave(all_plot, #path = prefix,
filename = paste0(pro, 'all_cut_point_survival_plot.pdf'),width = x2,height = y2)
因为它并不是中位值高低分组,所以两个分组的病人数量是不平衡的, 如下所示:
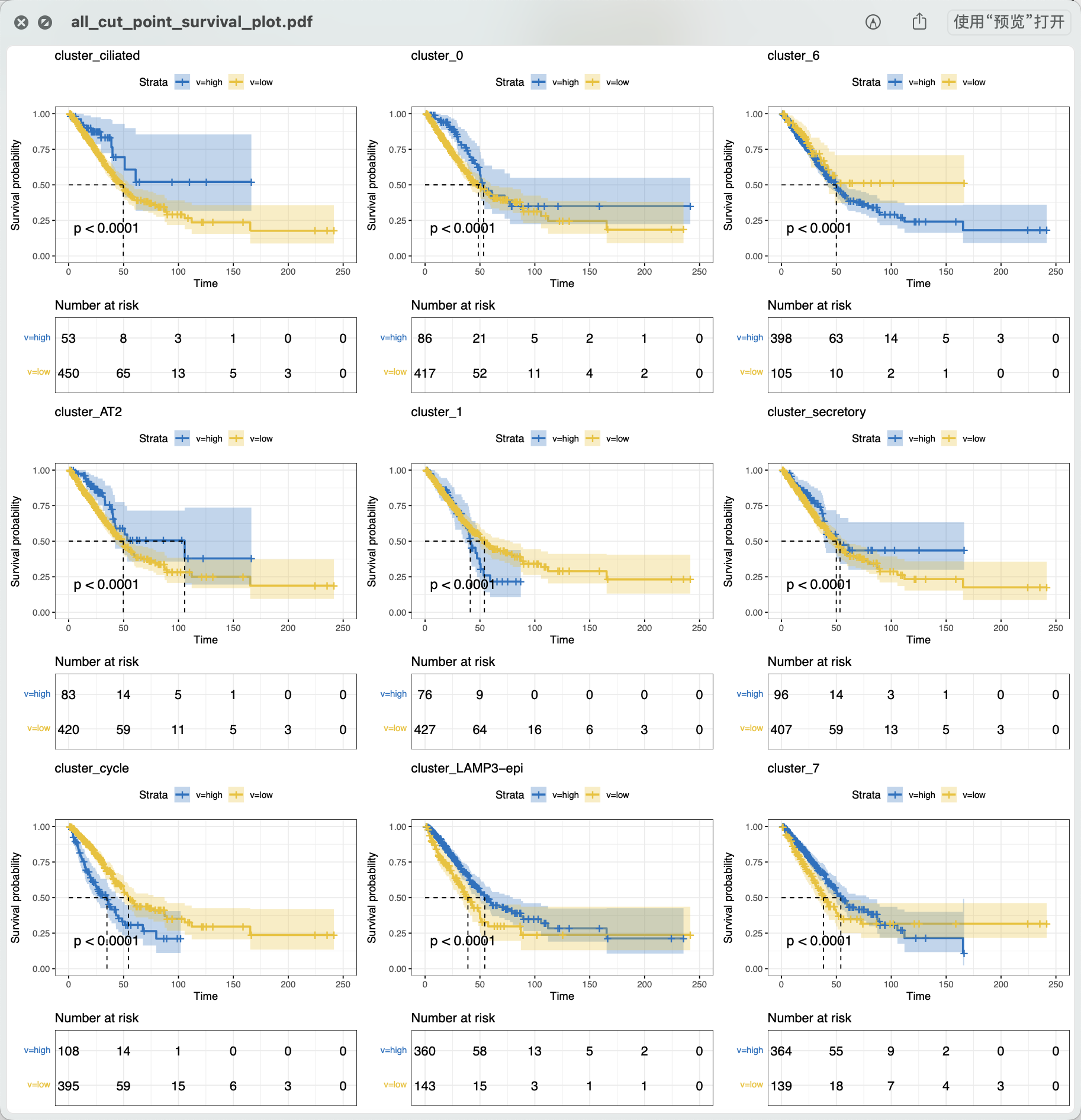
很明显可以看到,它取巧了,所以基本上可以让每个单细胞亚群的特异性基因列表,都有统计学显著的生存分析结果哦!
后记
我们按照文章的思路, 定位到了特殊的也就是具有统计学生存分析意义的亚群,详见:换一个分析策略会导致文章的全部论点都得推倒重来吗,但是也有其它思路可以拿到同样的结果。但是上面的代码已经是足够复杂啦,我们还是等待大家运行后我们先视频号答疑后,再分享后面的思路哈!代码会持续更新在这个链接哈,大家一定要抽空测试:
链接: https://pan.baidu.com/s/1geW1MTLRizcJWEESdjMN7g?pwd=96t6 提取码: 96t6
我们会在视频号直播互动答疑,欢迎预约我们的直播链接!