最近刷到了一个2023的文章《DNA methylome and transcriptome identified Key genes and pathways involved in Speckled Eggshell formation in aged laying hens》,家禽蛋壳的质量与蛋品生产的盈利能力密切相关。蛋壳斑点是影响鸡蛋外观和顾客偏好的一个重要品质性状。然而,斑点的形成机制仍然知之甚少。有斑点蛋和正常蛋的主要区别包括:
- 沉积物类型:
- 斑点蛋:蛋壳上的斑点可能是因为在蛋管中形成时,蛋壳上吸附了一些附加的色素或杂质,这可能包括血迹、食物残渣等。
- 正常蛋:正常蛋通常不包含这些外部沉积物,因此在外观上没有斑点。
- 形成机制:
- 斑点蛋:斑点的形成可能与鸡的生理状态、饮食、生活环境等因素有关。一些因素,如母鸡的健康状况、饲料中的色素含量、蛋壳表面的微小损伤等,都可能影响斑点的形成。
- 正常蛋:正常蛋通常在蛋管中形成时,蛋壳形成的过程相对均匀,没有明显的斑点。
- 外观和顾客偏好:
- 斑点蛋:由于斑点的存在,斑点蛋的外观可能会与正常蛋有所不同。有些消费者可能会对蛋壳斑点有不同的接受程度,有些人可能更喜欢无斑点的蛋。
- 正常蛋:正常蛋的外观相对均匀,更符合一些顾客的审美偏好。
肉眼确实是能分辨有斑点蛋和正常蛋:

所以研究者就针对有斑点蛋和正常蛋的whole-genome bisulfite sequencing (WGBS) and RNA-seq 数据,然后发现几乎没有差异,并且完全没有交集。。。
转录组测序后的差异分析
从质量控制可以看到, 有斑点蛋和正常蛋应该是在表达量的全局水平是没有分组差异的,如下所示:
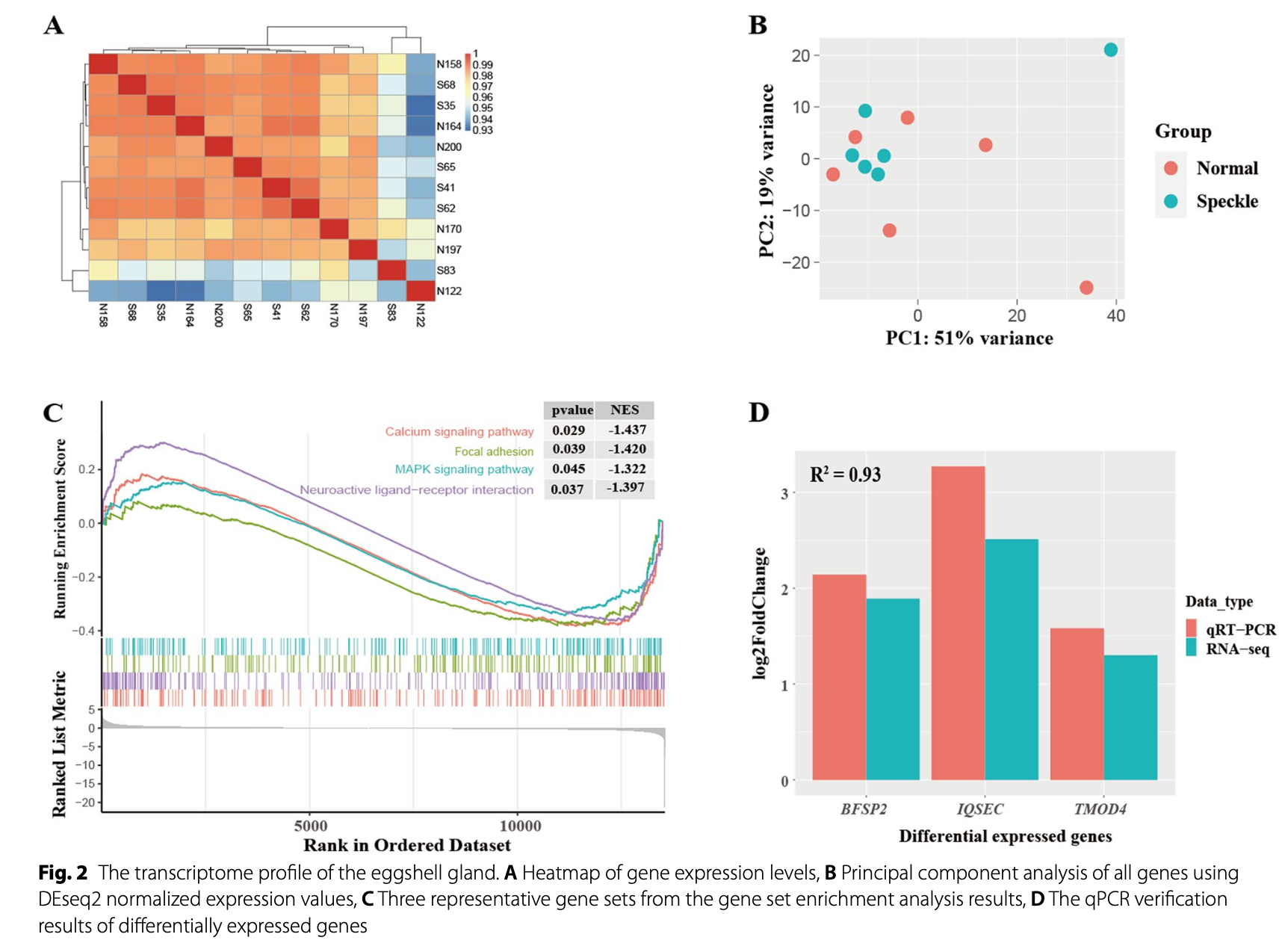
很明显就:
- RNA-seq analysis identified seven DEGs between the speckle and normal groups, includ- ing IQSEC3, BFSP2, TMOD4, LOC112530987, GABRA2, TRIQK, and a pseudogene. IQSEC3,
- Three DEGs (BFSP2, IQSEC, TMOD4) identified by RNA-seq were verified using quantitative real-time PCR
虽然说差异基因数量确实是很少,但是作者这里采取的是GSEA方法,并不是上下调基因各自的超几何分布检验的富集分析方法,所以并不会影响作者看具体的生物学通路变化。而且就个位数的差异基因居然可以通过湿实验验证出来3个。
与WGBS数据几乎没有交集
同样的,从质量控制可以看到, 两个分组的wgbs数据其实是有系统性的分组差异,所以有A total of 2788 differentially methylated regions (DMRs) were identified between the normal and speckle groups.
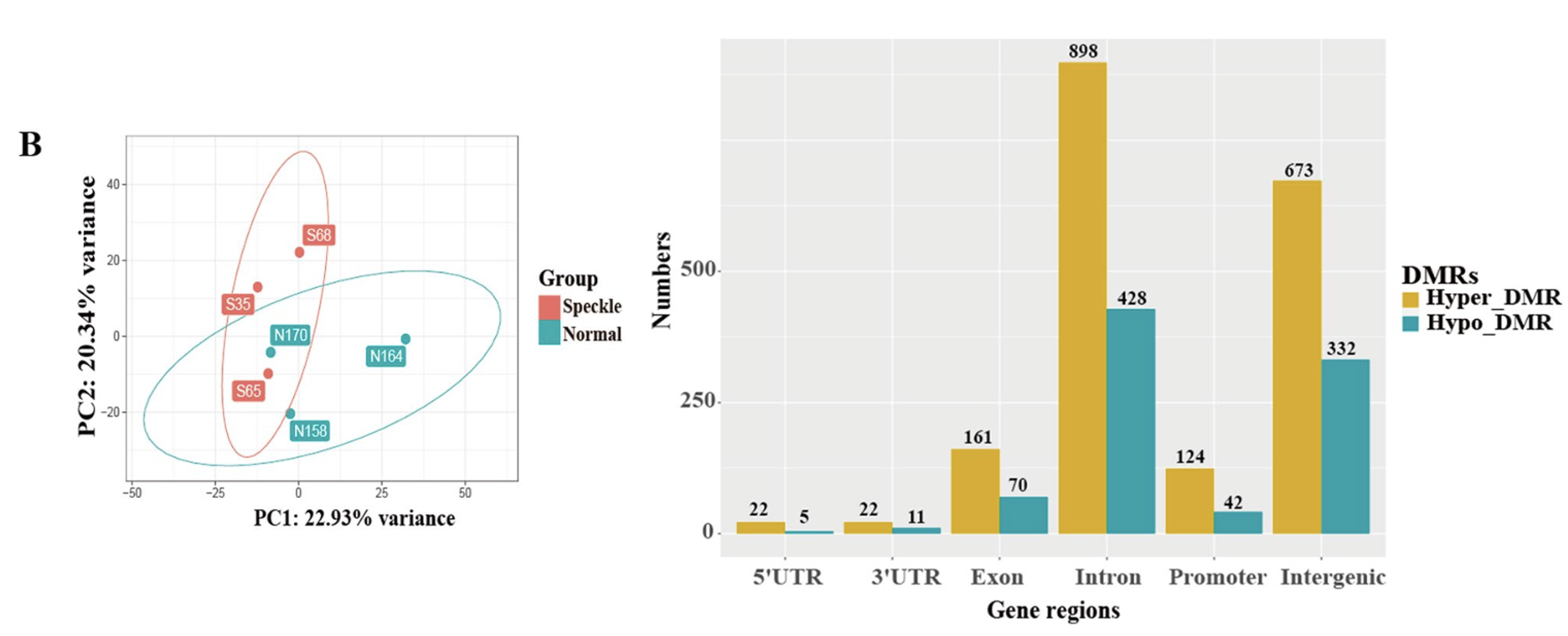
但是因为前面的转录组差异分析的目标基因数量实在是太少了,所以与WGBS数据几乎没有交集,如下所示:
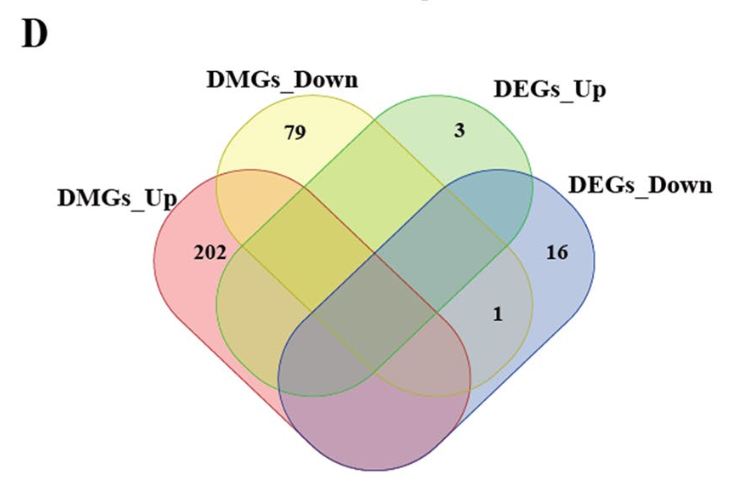
但是其实两个分组的转录组测序(RNA-Seq)和全基因组甲基化测序(WGBS)结果没有交集的情况可能涉及多个因素,以下是一些可能的解释:
- 功能独立:
- 转录组测序和全基因组甲基化测序测量的是细胞不同方面的生物学特征。如果这两者之间没有交集,可能是因为基因表达和DNA甲基化的调控机制在这两个过程中是相对独立的。
- 测序深度和灵敏度:
- 两种测序技术的深度和灵敏度不同,可能导致某些差异基因或甲基化位点未被检测到。确保测序深度足够,并且考虑到不同技术的灵敏度,可以增加两者之间的交集。
- 细胞类型异质性:
- 如果样本中存在细胞异质性,一种测序技术可能主要捕捉到某个亚群体的信号,而另一种技术可能捕捉到其他亚群体的信号。这可能导致两者之间的交集较小。
- 生物学变异:
- 不同的生物学过程可能在不同的条件下发生,因此导致差异基因和甲基化位点没有交集。例如,某些差异基因可能主要参与细胞信号传导,而甲基化变化可能主要发生在调控基因的启动子区域。
- 实验设计和条件选择:
- 如果实验设计中选择了不同的条件或不同的时间点,可能导致差异基因和甲基化位点在这两个实验中没有交集。
- 数据分析方法:
- 不同的数据分析方法可能导致不同的结果。确保采用合适的统计方法和分析流程可以减少假阳性和假阴性结果,增加两者之间的交集。
学徒作业
完成上面的文章的转录组测序(RNA-Seq)和全基因组甲基化测序(WGBS)的数据分析:
- DNA samples (n = 3 per group, Supplementary Table S1)
- RNA samples (n = 6 per group, Supplementary Table S1)
数据量并不大,很容易下载:
- https://www.ebi.ac.uk/ena/browser/view/PRJNA850950
- https://www.ebi.ac.uk/ena/browser/view/PRJNA851109
优先完成转录组测序(RNA-Seq)的定量和差异分析,作者得到如此少的差异基因数量也是让我很惊奇。
自己在Linux环境下面配置好转录组上游数据处理定量流程,然后对这个PRJNA850950的fq数据进行处理 ,首先需要 参考 在全新服务器配置转录组测序数据处理环境 ,主要是4个软件,如果安装成功后,下面的代码不会有error信息的:
- fastqc —help 1>/dev/null
- trim_galore —help 1>/dev/null
- hisat2 —help 1>/dev/null
- featureCounts —help 1>/dev/null
然后开启一个转录组实战,参考:https://mp.weixin.qq.com/s/YHWLcZYeKLEMufUS-TLHVQ
- 1.数据下载(自身数据的话,此步可忽略)
- 2.质控过滤(质控前用fastqc与multiqc初看数据效果、trimmgalore进行质控过滤与fastqc、multiqc查看质控后的效果)
- 3.Hisat2比对
- 4.featureCounts定量
拿到了表达量矩阵后走差异分析, 都是表达量矩阵而已,可以看到我8年前的芯片教程,推文在:
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
绘制差异基因的热图,以及火山图即可!