我们一直都有一个很简单的服务器共享业务,详见:生物信息学江湖的开创性产品-共享服务器。因为是同一个机器给很多人错峰使用,所以过去的五年一直运行的蛮好。
但是有一些代码本身有问题,所以也确实是有一些时候会造成整个服务器奔溃,比如一个小伙伴跑一个简单的动态预测模型代码,详见:
- 教程官网:https://drizopoulos.github.io/JMbayes2
- 本次教程地址:https://drizopoulos.github.io/JMbayes2/articles/Super_Learning.html
为了给他debug,他真实的数据的十几万条信息,为了简化,我们就直接使用了测试数据,代码很简单,就是加载 JMbayes2 包后使用里面的测试数据:
# install.packages('JMbayes2')
library(JMbayes2)
pbc2.id$status2 <- as.numeric(pbc2.id$status != 'alive')
CoxFit <- coxph(Surv(years, status2) ~ sex, data = pbc2.id)
fm1 <- lme(log(serBilir) ~ year * sex, data = pbc2, random = ~ year | id)
jointFit1 <- jm(CoxFit, fm1, time_var = "year",n_chains = 3L, cores =8L)
summary(jointFit1)
jointFit1$control
以下是代码的解释:
library(JMbayes2): 加载JMbayes2包,该包提供了执行联合模型分析的功能。pbc2.id$status2 <- as.numeric(pbc2.id$status != 'alive'): 创建一个新的变量status2,其值为 1 或 0,表示生存状态。通常,1 表示死亡,0 表示存活。CoxFit <- coxph(Surv(years, status2) ~ sex, data = pbc2.id): 使用 Cox 比例风险模型拟合生存数据。这个模型通过coxph函数构建,其中Surv(years, status2)指定了生存时间和状态,而sex是预测变量。fm1 <- lme(log(serBilir) ~ year * sex, data = pbc2, random = ~ year | id): 创建一个混合效应模型,使用lme函数。这个模型是关于log(serBilir)的,其中year和sex是预测变量,而id是随机效应。jointFit1 <- jm(CoxFit, fm1, time_var = "year", n_chains = 3L, cores = 8L): 将 Cox 模型和混合效应模型联合起来。这个联合模型使用jm函数创建,其中CoxFit和fm1是先前创建的模型,time_var = "year"指定时间变量。summary(jointFit1): 打印联合模型的摘要信息,包括各个模型的参数估计和统计检验等。jointFit1$control: 这是联合模型的控制参数,包含模型拟合时使用的一些设置。可以查看这些参数以了解模型拟合的细节。
这个 JMbayes2 包的测试集比较简单,如下所示:
> head(pbc2.id[,1:6])
id years status drug age sex
1 1 1.095170 dead D-penicil 58.76684 female
2 2 14.152338 alive D-penicil 56.44782 female
3 3 2.770781 dead D-penicil 70.07447 male
4 4 5.270507 dead D-penicil 54.74209 female
5 5 4.120578 transplanted placebo 38.10645 female
6 6 6.853028 dead placebo 66.26054 female
> dim(pbc2.id)
[1] 312 20
> colnames(pbc2.id)
[1] "id" "years" "status"
[4] "drug" "age" "sex"
[7] "year" "ascites" "hepatomegaly"
[10] "spiders" "edema" "serBilir"
[13] "serChol" "albumin" "alkaline"
[16] "SGOT" "platelets" "prothrombin"
[19] "histologic" "status2"
如果是把上面的312行的数据扩充到十几万条信息然后跑上面的代码,可能是确实是需要服务器。这个测试数据,在我们个人电脑通常是十几秒钟就跑完了,但是如果是服务器的话居然是需要十几分钟,就很让用户奔溃,关键是慢就算了还会把服务器卡死,因为调用了服务器所有的计算机资源。。。。
首先我们打开了哪个卡死服务器的函数的代码本身,看了看里面关于线程的细节:
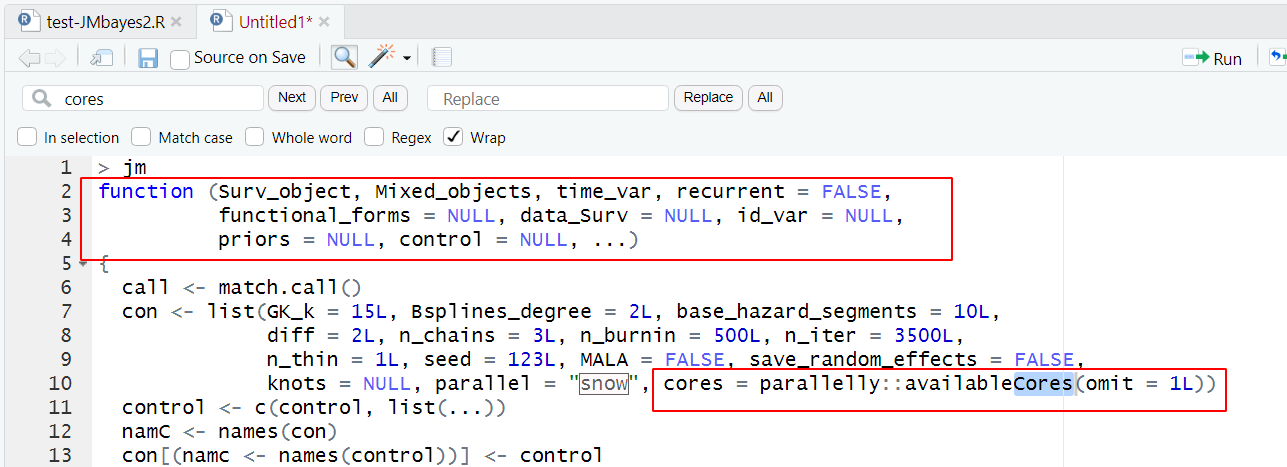
也就是说,我们如果使用这个函数的时候人为的传输参数设置就使用少量的几个线程其实没有意义,因为这个函数压根就不会解析我们的传入进去的参数。而且我们去看了看这个包的github源代码,确实是这样的:
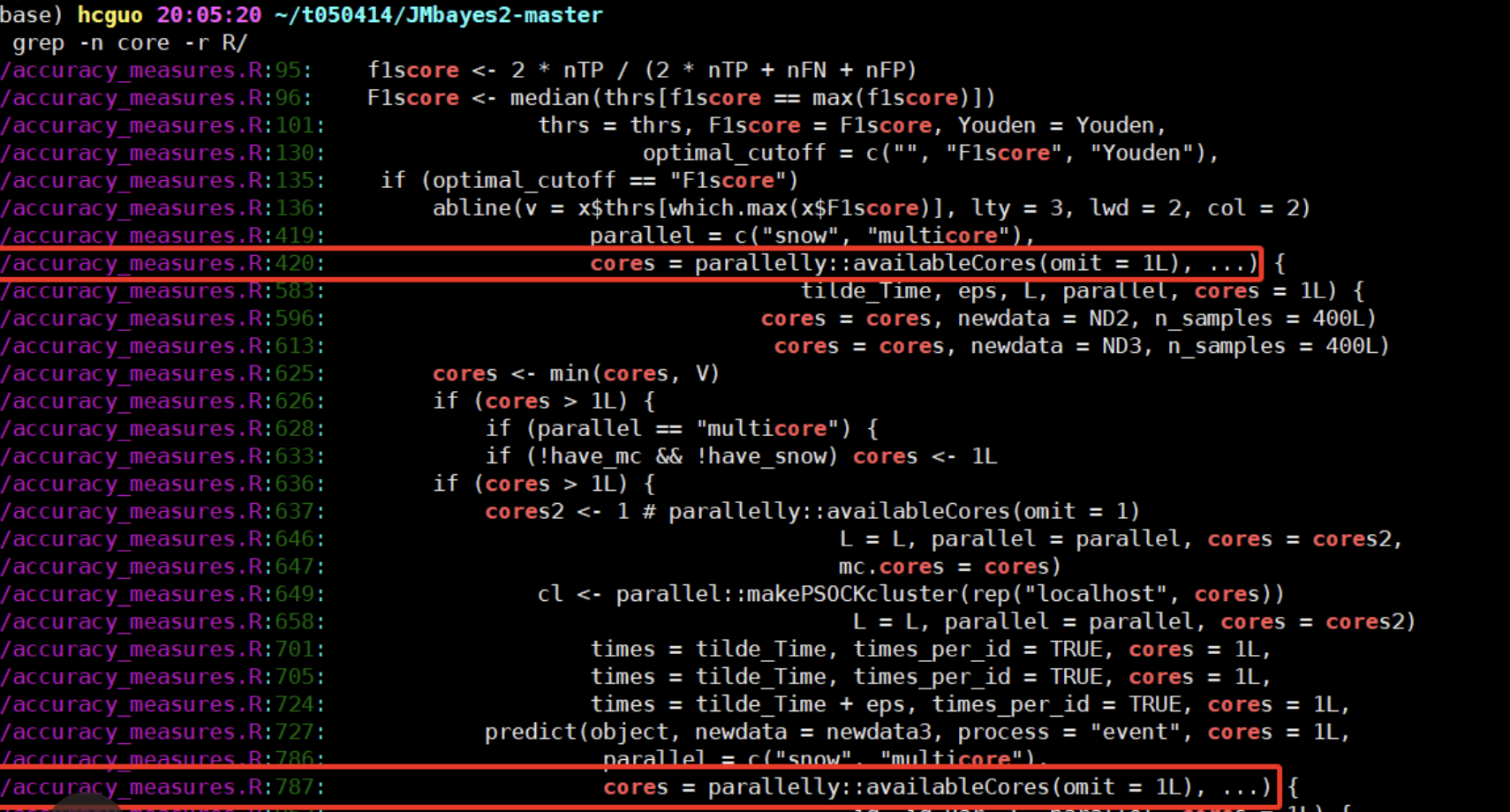
也就是说作者选择了函数可以自动检测可用的处理器核心数,并根据可用核心数进行多线程处理。这样可以充分利用计算资源,而不需要用户手动设置。
my_function <- function(...) {
num_threads <- parallel::detectCores()
# 在需要时使用 num_threads 控制多线程的逻辑
# 具体的多线程处理代码
}
并没有真实的接受用户传入参数,正常情况下应该是真实的允许用户在调用函数时手动设置线程数,这样用户可以根据实际需求选择是否使用多线程。如果用户没有指定线程数,函数可以使用默认值。
my_function <- function(..., num_threads = 1) {
# 在需要时使用 num_threads 控制多线程的逻辑
# 具体的多线程处理代码
}
我们的服务器动辄是256个线程
详见生物信息学江湖的开创性产品-共享服务器,也就是说上面的代码如果是自动检测服务器可用线程,那就很夸张了,开启如此多的线程本来就是很耗费时间的,而且对计算机资源的消耗更棘手。