前面我们在推文:数据分析有错误并不可怕,造假才不可饶恕 提到了这个新鲜出炉( 2023年12月5日)的cell期刊的文章单细胞转录组数据分析环节是有一些瑕疵的。
首先是在 单细胞水平这样的细胞比例变化可靠吗 已经提到了两个分组的单细胞亚群比例变化问题,很大程度上受到了离群点的影响。另外就是总体上这个cell期刊的文章的降维聚类分群后的拿到的各个亚群的特异性高表达量的基因列表就不常见,但是如果仅仅是使用作者提供的矩阵文件那么就很难搞清楚问题出在哪里,所以我们还是从单细胞转录组的测序数据开始:
- https://www.ncbi.nlm.nih.gov/bioproject/PRJNA856132
- https://ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE207546
可以看到,里面确实是15个样品,分别是常规转录组,单细胞转录组,以及空间单细胞转录组,如下所示:
- 单细胞核转录组的6个小鼠样品,sn-seq: two exprimental groups: normal and EO771-VM-bearing mice, 3 independent replicates with 5 mice per group in each replicate.
- 空间单细胞转录组的3个样品,ST, 3 brain sections from 3 EO771-VM mice.
- 人类细胞系的6个样品,RNA-seq: human choroid plexus epithelial cell line (HCPEpiC) treated with or without tryptase (3 independent replicates per group).
下面的内容会涉及到:
- 学习cellranger的定量流程
- 从零开始在自己的服务器安装软件
- 下载测序数据
- 走流程
cellranger的定量流程详解:
正常走cellranger的定量流程即可,代码我已经是多次分享了。参考:
- 10X单细胞转录组原始测序数据的Cell Ranger流程(仅需800元)
- 10X的单细胞转录组原始数据也可以在EBI下载
- 一个10x单细胞转录组项目从fastq到细胞亚群
- 一文打通单细胞上游:从软件部署到上游分析
- PRJNA713302这个10x单细胞fastq实战
- 一次曲折且昂贵的单细胞公共数据获取与上游处理
- 只能下载bam文件的10x单细胞转录组项目数据处理
- 不知道10x单细胞转录组样品和fastq文件的对应关系
- 10X单细胞转录组测序数据的 SRA转fastq踩坑那些事
- 10x的单细胞转录组fastq文件的R1和R2不能弄混哦
差不多几个小时就可以完成全部的样品的cellranger的定量流程。基础知识非常重要,我们在单细胞天地多次分享过cellranger流程的笔记(2019年5月),大家可以自行前往学习,如下:
- 单细胞实战(一)数据下载
- 单细胞实战(二) cell ranger使用前注意事项
- 单细胞实战(三) Cell Ranger使用初探
- 单细胞实战(四) Cell Ranger流程概览
- 单细胞实战(五) 理解cellranger count的结果
安装自己的conda
在自己的服务器里面自己下载并且安装自己的conda,自己配置哈:
# 首先下载文件,20M/S的话需要几秒钟即可
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
# Miniconda3-latest-Linux-x86_64.sh 135.1 MiB 2023-12-21 09:23
# wget https://mirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/Miniconda3-latest-Linux-x86_64.sh
# 如果比较慢就切换镜像哦
# 接下来使用bash命令来运行我们下载的文件,记得是一路yes下去
bash Miniconda3-latest-Linux-x86_64.sh
# 上面的bash命令安装成功后,需要更新系统环境变量文件
source ~/.bashrc
首先如果是在中国大陆,需要设置好镜像:
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/cloud/bioconda/
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/pkgs/free/
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/pkgs/main/
conda config --set show_channel_urls yes
然后就可以使用自己的conda来安装 kingfisher 软件:
conda install mamba
mamba create -n kingfisher python=3.8
mamba init
mamba list
# 首先重启terminal,然后使用下面的命令 :
mamba activate kingfisher
mamba install -c bioconda kingfisher
然后就一行代码下载数据,示例代码如下所示:
kingfisher get -p PRJNA853539 -m ena-ascp ena-ftp prefetch aws-http
另外,值得注意的是,因为上面的代码耗时很久,一般来说需要使用nohup包裹一下,或者说开启screen命令。
使用kingfisher下载测序数据
因为我们前面拿到了测序数据相关信息:
- https://www.ncbi.nlm.nih.gov/bioproject/PRJNA856132
- https://ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE207546
所以我们本次的一行代码下载数据应该是如下所示:
nohup kingfisher get -p PRJNA856132 -m ena-ascp ena-ftp prefetch aws-http &
# 01/20/2024 04:07:00 PM INFO: Kingfisher v0.3.1
# 01/23/2024 02:11:04 PM INFO: Kingfisher v0.4.1
但是因为这个代码过于简单,所以它其实是在某些场合会报错,比如这个数据集就是里面有人类和小鼠数据混搭,而且人类细胞系数据呢在ena数据库里面并没有,但是在ncbi的sra数据库是有的!所以其实是可以分开下载,比如我们首先挑选小鼠数据,代码如下所示:
# 首先获取项目的全部的样品测序信息
kingfisher annotate -p PRJNA856132 -f csv --all-column -o ./annotate_info.csv
# 从里面挑选出来小鼠相关的数据
$cat annotate_info.csv |awk -F "," 'NR>1{if($8=="Mus musculus"){print $1 "\t" $8}}'
SRR19996402 Mus musculus
SRR19996403 Mus musculus
SRR19996404 Mus musculus
SRR19996405 Mus musculus
SRR19996406 Mus musculus
SRR19996407 Mus musculus
SRR19996408 Mus musculus
cat annotate_info.csv |awk -F "," 'NR>1{if($8=="Mus musculus"){print $1 "\t" $8}}'|cut -f 1 > SRR_list.txt
screen -r kinf
/usr/bin/time -v kingfisher get --run-identifiers-list ./sample_info/SRR_list.txt -m ena-ascp ena-ftp prefetch --download-threads 10 --check-md5sums 1>down_srr_list.log 2>&1
这样的话,开启screen命令后,就可以使用kingfisher慢慢的下载我们的所需要的小鼠相关测序数据啦。
如下所示的全部的测序的fq文件:
ls -lh SRR*gz |cut -d" " -f 5- |head -20
15G 1月 12 18:20 SRR19996402_1.fastq.gz
17G 1月 12 18:31 SRR19996402_2.fastq.gz
21G 1月 14 12:36 SRR19996403_1.fastq.gz
26G 1月 14 12:54 SRR19996403_2.fastq.gz
14G 1月 14 18:23 SRR19996404_1.fastq.gz
15G 1月 14 18:32 SRR19996404_2.fastq.gz
511M 1月 12 19:24 SRR19996405_1.fastq.gz
320M 1月 12 19:25 SRR19996405_2.fastq.gz
572M 1月 12 19:29 SRR19996406_1.fastq.gz
359M 1月 12 19:30 SRR19996406_2.fastq.gz
644M 1月 14 17:09 SRR19996407_1.fastq.gz
554M 1月 14 17:10 SRR19996407_2.fastq.gz
528M 1月 12 19:34 SRR19996408_1.fastq.gz
453M 1月 12 19:34 SRR19996408_2.fastq.gz
624M 1月 12 19:36 SRR19996409_1.fastq.gz
415M 1月 12 19:37 SRR19996409_2.fastq.gz
595M 1月 12 19:38 SRR19996410_1.fastq.gz
396M 1月 12 19:39 SRR19996410_2.fastq.gz
1.2G 1月 12 19:43 SRR19996411_1.fastq.gz
880M 1月 12 19:44 SRR19996411_2.fastq.gz
........ ........ ........ ........ ........
cellranger的定量流程首先需要合理的文件名字
上面下载的全部的测序的fq文件名字是srr开头的,我们样品名字是gsm开头的, 两码事,需要有对应关系!一个简单的Linux命令就可以获取他们的对应关系
cat annotate_info.csv |awk -F "," 'NR>1{if($8=="Mus musculus"){print $1 "\t" $10}}' |awk -F "\t|:|;" '{print $1 "\t" $2 "\t"$3}'
SRR19996402 GSM6294364 ST3
SRR19996403 GSM6294363 ST2
SRR19996404 GSM6294362 ST1
SRR19996405 GSM6294361 treatment3
SRR19996406 GSM6294361 treatment3
SRR19996407 GSM6294358 normal3
SRR19996408 GSM6294358 normal3
SRR19996409 GSM6294360 treatment2
SRR19996410 GSM6294360 treatment2
SRR19996411 GSM6294360 treatment2
........ ........ ........
有了这样的对应关系,就比较好改名啦,如果你熟悉10x单细胞转录组数据,就知道:
- 首先,1-26个cycle就是测序得到了26个碱基,先是16个Barcode碱基,然后是10个UMI碱基;通常是R1文件
- 然后,27-34这8个cycle得到了8个碱基,就是i7的sample index;通常是I1文件
- 最后35-132个cycle得到了98个碱基,就是转录本reads(目前很多测序仪都是150bp了),通常是R2文件
也就是说R2 文件是真正的测序reads,肯定是文件比较大哦,但是我们前面看到的r1和r2文件其实很难看出来文件大小差距,因为里面的测序数据并没有被切除成为规定的碱基长度,这个并不会影响我们的cellranger的定量流程。
因为每一个gsm的样品会对应多个srr的id编号,所以这个文件名修改会比较复杂。我们这里直接给大家最终代码让大家看看,其中的两个gsm的样品如下所示:
$cat change_R1.sh
ln -s SRR19996442_1.fastq.gz GSM6294356_normal1_S1_L001_R1_001.fastq.gz
ln -s SRR19996443_1.fastq.gz GSM6294356_normal1_S2_L001_R1_001.fastq.gz
ln -s SRR19996444_1.fastq.gz GSM6294356_normal1_S3_L001_R1_001.fastq.gz
ln -s SRR19996445_1.fastq.gz GSM6294356_normal1_S4_L001_R1_001.fastq.gz
ln -s SRR19996446_1.fastq.gz GSM6294356_normal1_S5_L001_R1_001.fastq.gz
ln -s SRR19996447_1.fastq.gz GSM6294356_normal1_S6_L001_R1_001.fastq.gz
ln -s SRR19996448_1.fastq.gz GSM6294356_normal1_S7_L001_R1_001.fastq.gz
ln -s SRR19996449_1.fastq.gz GSM6294356_normal1_S8_L001_R1_001.fastq.gz
ln -s SRR19996431_1.fastq.gz GSM6294357_normal2_S1_L001_R1_001.fastq.gz
ln -s SRR19996432_1.fastq.gz GSM6294357_normal2_S2_L001_R1_001.fastq.gz
ln -s SRR19996433_1.fastq.gz GSM6294357_normal2_S3_L001_R1_001.fastq.gz
ln -s SRR19996434_1.fastq.gz GSM6294357_normal2_S4_L001_R1_001.fastq.gz
ln -s SRR19996435_1.fastq.gz GSM6294357_normal2_S5_L001_R1_001.fastq.gz
ln -s SRR19996450_1.fastq.gz GSM6294357_normal2_S6_L001_R1_001.fastq.gz
ln -s SRR19996451_1.fastq.gz GSM6294357_normal2_S7_L001_R1_001.fastq.gz
ln -s SRR19996452_1.fastq.gz GSM6294357_normal2_S8_L001_R1_001.fastq.gz
可以看到这些id对应关系并不是顺序编号的,具体大家可以各显神通,自己摸索这个过程哈 。
然后正常配置cellranger的定量流程
理论上应该是去10x的官网下载cellranger软件和数据库文件,不过我们的共享服务器已经给大家准备了,在 /home/data/refdir/10x 文件夹里面:
ls -lh /home/data/refdir/10x |cut -d" " -f 5-
391M 8月 8 2022 cellranger-6.0.1.tar.gz
11G 8月 7 2022 refdata-gex-GRCh38-2020-A.tar.gz
9.9G 8月 7 2022 refdata-gex-GRCh38-and-mm10-2020-A.tar.gz
9.7G 8月 7 2022 refdata-gex-mm10-2020-A.tar.gz
在Linux中,你可以使用tar命令来解压一个.tar.gz文件,并将解压后的文件放到指定的文件夹。以下是具体的命令:
tar -xzvf file.tar.gz -C /path/to/directory
在这个命令中:
x代表解压z代表gzip,表示文件是gzip格式的,需要进行解压v代表verbose,表示显示详细的解压过程f代表file,表示后面跟的是要解压的文件名-C代表change to directory,表示解压后的文件将被放到后面指定的目录
请将file.tar.gz替换为你的文件名,将/path/to/directory替换为你想要放置解压后文件的目录。
那么,我们的命令就是:
mkdir raw run soft
mv *gz raw
tar -xzvf /home/data/refdir/10x/cellranger-6.0.1.tar.gz -C ./soft
tar -xzvf /home/data/refdir/10x/refdata-gex-mm10-2020-A.tar.gz -C ./soft
值得注意的是cellranger是一直在更新的,所以推荐去官网下载最新版,这里cellranger的下载链接会失效 ,需要去官网获取:https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest
有了10x的官网下载cellranger软件和数据库文件,以及前面的kingfisher下载了的小鼠的6个样品的10x技术单细胞转录组上游测序数据,接下来就可以跑cellranger啦,因为是共享服务器,如果不着急的话,可以一个个样品慢慢跑代码。也可以5个样品一起运行:
ref=../soft/refdata-gex-mm10-2020-A/
cellranger=../soft/cellranger-7.1.0/cellranger
# ls GSM*.fastq.gz
ls ../raw/*gz|cut -d"_" -f 1 |sort -u|cut -d"/" -f 3 | cut -d "_" -f 1 | uniq | while read id;do
nohup $cellranger count --id=$id \
--transcriptome=$ref \
--fastqs=../raw \
--sample=$id \
--nosecondary \
--localcores=4 \
--localmem=30 &
done
因为是5个样品一起运行,所以每个样品耗时三五个小时,合起来也是四五个小时就足够啦。虽然说5个样品一起运行会很快,但是大家的CPU和内存可能会不够,如果是共享服务器或者云服务器,另外一个门槛是硬盘。。。。
有可能是硬盘也不够哦。。。。如果是这样的话,上面的代码大家千万不要添加 nohup ,应该是把上面的代码写入脚本后一个个运行。然后简单的整理一下结果矩阵和报表即可:
pro=outputs
pro_folder=$pro
mkdir -p $pro_folder
ls -lh $pro_folder
mkdir -p $pro_folder/html
ls */outs/web_summary.html |while read id;do (cp $id $pro_folder/html/${id%%/*}.html );done
mkdir -p $pro_folder/matrix
ls -d */outs/filtered_feature_bc_matrix |while read id;do (cp -r $id $pro_folder/matrix/${id%%/*} );done
很容易拿到了小鼠的6个样品的10x技术单细胞转录组上游定量后的表达量矩阵文件和报表,如下所示:
$ tree -h PRJNA856132_outs/
PRJNA856132_outs/
├── [4.0K] html
│ ├── [3.2M] GSM6294356_normal1.html
│ ├── [3.2M] GSM6294357_normal2.html
│ ├── [3.0M] GSM6294358_normal3.html
│ ├── [2.7M] GSM6294359_treatment1.html
│ ├── [2.6M] GSM6294360_treatment2.html
│ └── [2.9M] GSM6294361_treatment3.html
├── [4.0K] matrix
│ ├── [4.0K] GSM6294356_normal1
│ │ ├── [ 20K] barcodes.tsv.gz
│ │ ├── [284K] features.tsv.gz
│ │ └── [ 11M] matrix.mtx.gz
│ ├── [4.0K] GSM6294357_normal2
│ │ ├── [ 23K] barcodes.tsv.gz
│ │ ├── [284K] features.tsv.gz
│ │ └── [ 16M] matrix.mtx.gz
│ ├── [4.0K] GSM6294358_normal3
│ │ ├── [ 21K] barcodes.tsv.gz
│ │ ├── [284K] features.tsv.gz
│ │ └── [ 12M] matrix.mtx.gz
│ ├── [4.0K] GSM6294359_treatment1
│ │ ├── [ 24K] barcodes.tsv.gz
│ │ ├── [284K] features.tsv.gz
│ │ └── [8.5M] matrix.mtx.gz
│ ├── [4.0K] GSM6294360_treatment2
│ │ ├── [ 14K] barcodes.tsv.gz
│ │ ├── [284K] features.tsv.gz
│ │ └── [2.5M] matrix.mtx.gz
│ └── [4.0K] GSM6294361_treatment3
│ ├── [ 20K] barcodes.tsv.gz
│ ├── [284K] features.tsv.gz
│ └── [4.3M] matrix.mtx.gz
└── [ 61M] PRJNA856132_outs.tar.gz
8 directories, 25 files
接下来我们就可以针对这个文件夹(PRJNA856132_outs/matrix )走我们的单细胞转录组降维聚类分群流程啦。
先看看定量结果
并不是说每个样品质量都不行,起码这个 GSM6294356_normal1 还算是勉强可以的,虽然说中位数的基因确实是有点寒碜,以及右边的曲线真的是一言难尽啊:
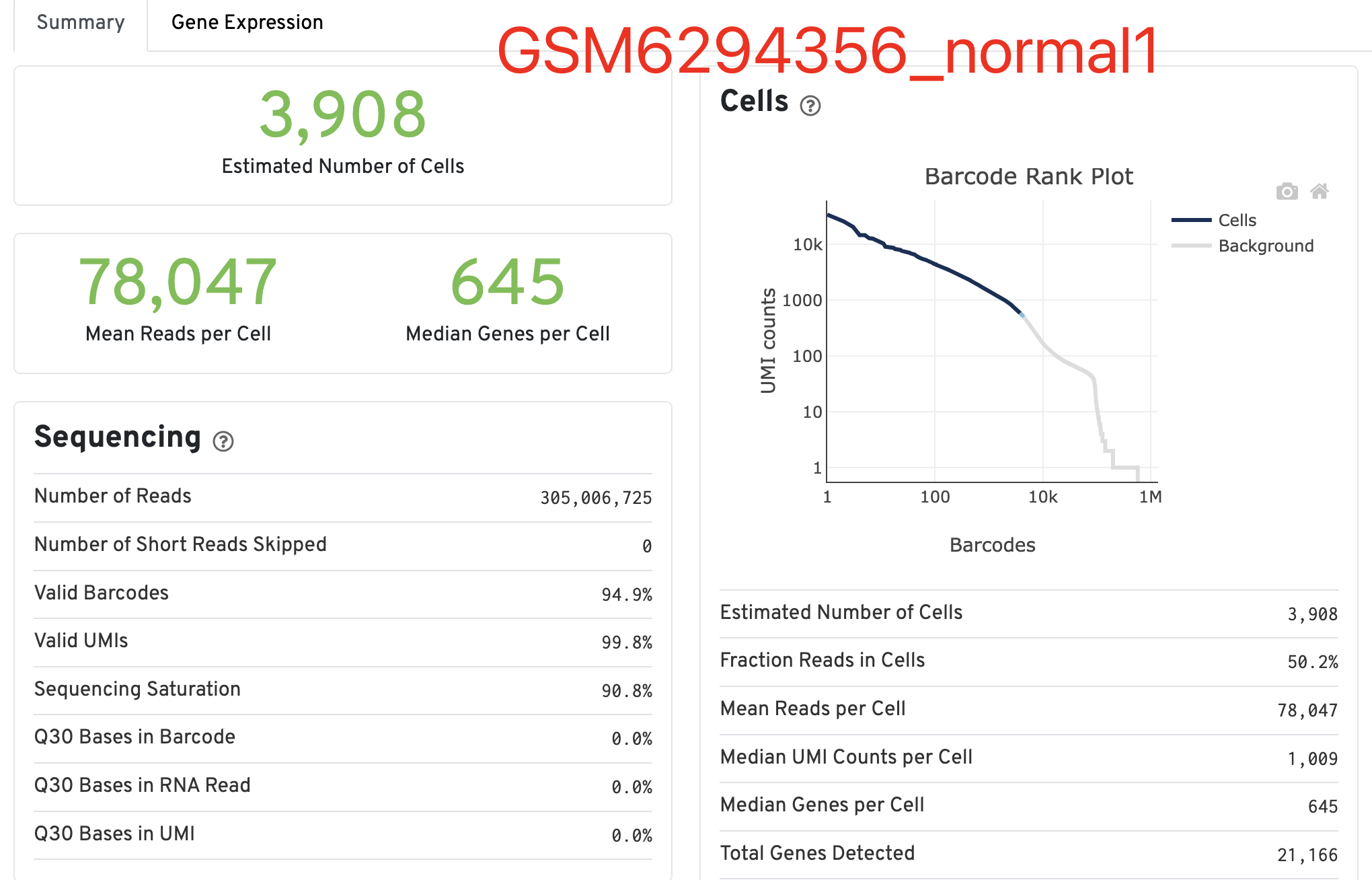
那么, 质量差的能到什么程度呢?让我们来看看这个GSM6294361_treatment3样品吧,我都不能理解,看到了这样的数据分析报表难道不应该是直接放弃这批样品吗?
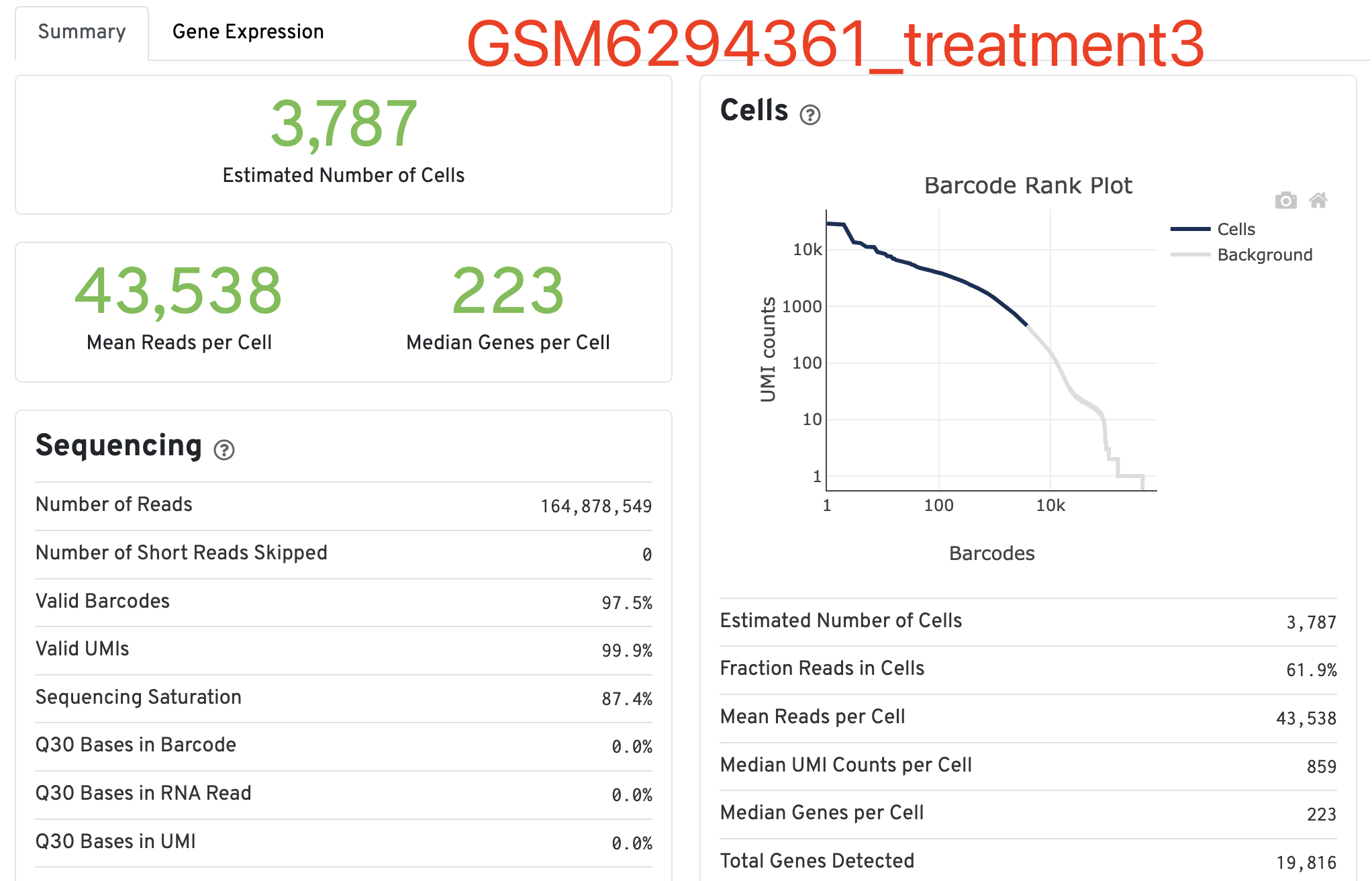
如果这个新鲜出炉( 2023年12月5日)的cell期刊的文章,需要靠这样的质量堪忧的单细胞转录组数据,那么它里面的生物学结论真的是值得信赖吗?
再次求助神通广大的网友们
文章的数据都是公开的,我相信对大家来说处理起来并不难,我们应该是抱着求知若渴的态度认真剖析一些这个单细胞转录组数据吧:
- https://www.ncbi.nlm.nih.gov/bioproject/PRJNA856132
- https://ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE207546
我现在有了自己定量的单细胞转录组表达量矩阵文件(来源于PRJNA856132),也有了作者给出来的表达量矩阵文件(来源于GSE207546),接下来我们就把这两个结果都各自独立的分析去看看是否有什么冲突吧。