前面我们提到了空间单细胞约等于10x技术,就比较方便理解,虽然说也有其它空间单细胞技术可以产出各式各样的数据。详见:10x的空间单细胞文件格式详解
但是对初学者来说,重要的是如何把不同技术产出的表达量矩阵导入到R或者Python这样的编程语言环境里面。今天我们来介绍的是在R语言里面的最流行的Seurat的单细胞流程,第一步就是理解Seurat的空间单细胞对象结构。值得注意的是我们接下来(2023年12月30日之后)的教程都是基于Seurat的V5版本哦:
示范数据(10x技术)
来源于:https://satijalab.org/seurat/articles/spatial_vignette.html#x-visium
因为我们要介绍的是Seurat的空间单细胞对象结构,所以理论上使用Seurat的官方数据是最好的,但是它依赖于一个github包,对很多人来说因为在中国大陆地区,所以访问它会有一点点难度。
首先安装这个SeuratData包,它在satijalab的github上面,代码如下所示:
library(devtools) #本地安装需要devtools
devtools::install_github('satijalab/seurat-data')
library(SeuratData)
如果因为在中国大陆地区上面的代码失败了,就需要想办法找人帮你下载上面的seurat-data离线本地安装包然后本地安装也是一样的效果哈。
然后有了SeuratData包, 就可以使用它来安装任意示例数据集啦。
library(SeuratData)
options(timeout=100000)
AvailableData()
InstallData("stxBrain")
# https://github.com/satijalab/seurat-data
# trying URL 'http://seurat.nygenome.org/src/contrib/stxBrain.SeuratData_0.1.1.tar.gz'
# Content type 'application/octet-stream' length 110346050 bytes (105.2 MB)
上面的代码其实就是参考 https://github.com/satijalab/seurat-data 的代码后给你去 http://seurat.nygenome.org/ 下载一个 离线本地安装包然后本地安装也是一样的效果哈,比如上面的 stxBrain.SeuratData_0.1.1.tar.gz ,因为有 (105.2 MB),如果你在中国大陆地区上,那么面的代码失败也是很正常的。
安装示例数据成功之后就可以载入它然后查看这个,代码如下所示:
brain <- LoadData("stxBrain", type = "anterior1")
brain
> brain
An object of class Seurat
31053 features across 2696 samples within 1 assay
Active assay: Spatial (31053 features, 0 variable features)
2 layers present: counts, data
1 image present: anterior1
上面的示例数据是31053个基因在2696个样品(10个左右的细胞,spot等)里面的表达量矩阵的空间单细胞数据。
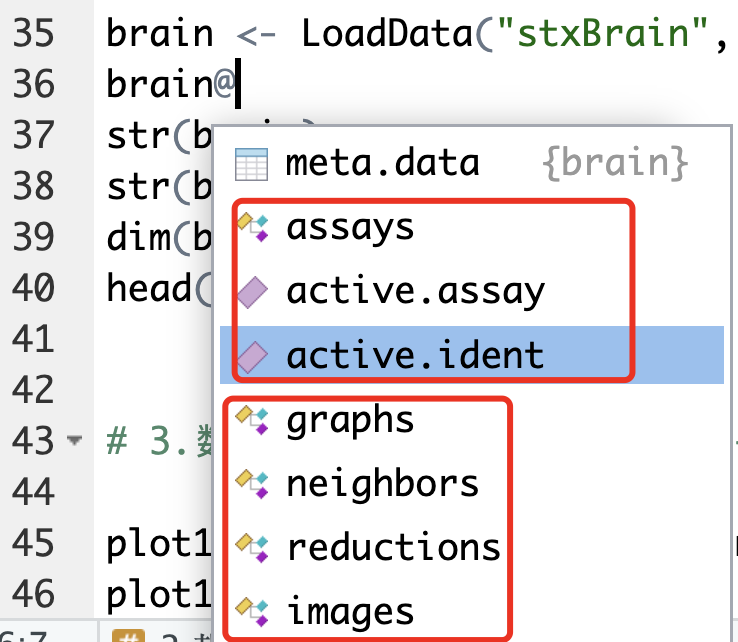
它里面的自带了一个图片信息, 一个表达量矩阵哦。简单的看看里面的结构发现第一层就有13个子对象,我们可以一个个来看看
> str(brain)
Formal class 'Seurat' [package "SeuratObject"] with 13 slots
..@ assays :List of 1
首先看对象里面的每个sample的属性(meta.data元素)
> dim(brain@meta.data)
[1] 2696 5
> head(brain@meta.data)
orig.ident nCount_Spatial nFeature_Spatial slice
AAACAAGTATCTCCCA-1 anterior1 13069 4242 1
AAACACCAATAACTGC-1 anterior1 37448 7860 1
AAACAGAGCGACTCCT-1 anterior1 28475 6332 1
AAACAGCTTTCAGAAG-1 anterior1 39718 7957 1
AAACAGGGTCTATATT-1 anterior1 33392 7791 1
AAACATGGTGAGAGGA-1 anterior1 20955 6291 1
对这个meta.data元素可以做各种各样的统计,因为它就是一个普通的数据框而已。
然后看表达量矩阵(assays相关元素)
它里面存储着最原始的表达量矩阵,以及不同算法处理过程的各种中间过程矩阵
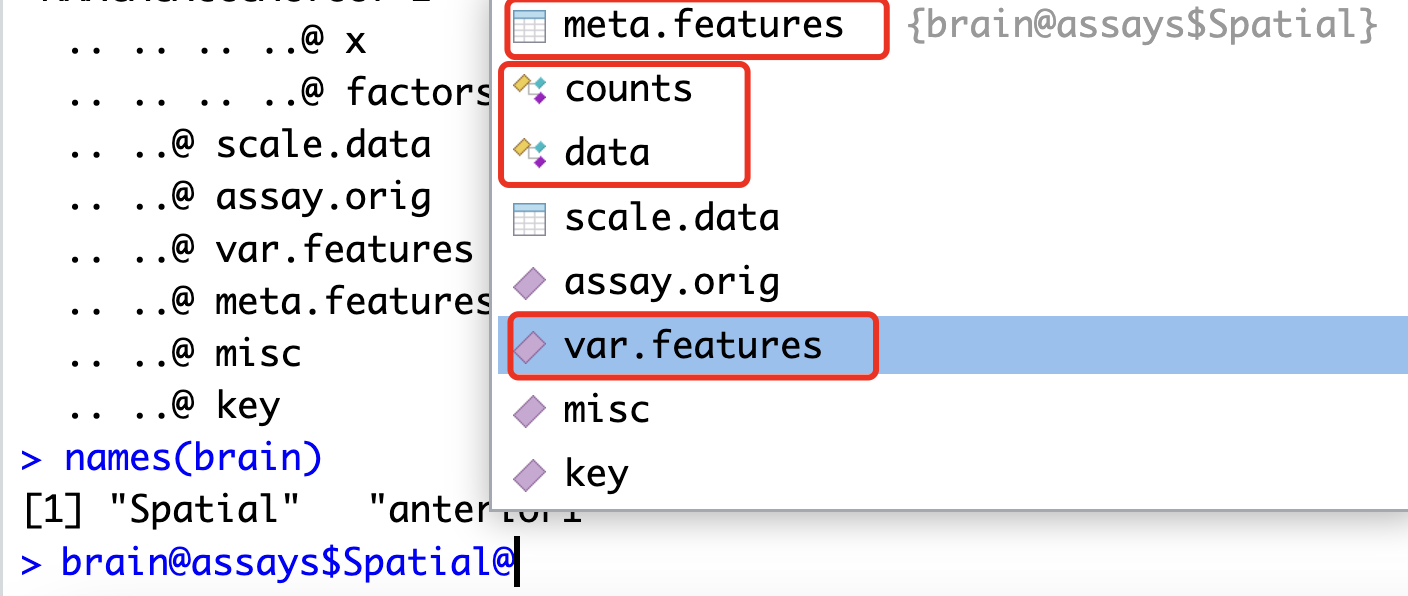
另外就是,如果经过了SCT的算法处理,会在assays下面多一个:
brain <- SCTransform(brain, assay = "Spatial", verbose = FALSE)
#它构建基因表达的正则化负二项式模型,以便在保留生物学差异的同时考虑技术伪影
> brain@assays$SCT
SCTAssay data with 17668 features for 2696 cells, and 1 SCTModel(s)
Top 10 variable features:
Ttr, Hbb-bs, Mbp, Plp1, Hba-a1, Ptgds, Penk, Hba-a2, S100a5,
Fabp7
目前看到的绝大部分文档都是说SCT的算法比较适合空间单细胞转录组的表达量矩阵的处理哦。
然后看看空间信息(images元素)
它里面存储的全部的空间信息,里面的元素也可以打开一个个探索:

比如样品的空间切片坐标信息:
> head(brain@images$anterior1@coordinates)
tissue row col imagerow imagecol
AAACAAGTATCTCCCA-1 1 50 102 7475 8501
AAACACCAATAACTGC-1 1 59 19 8553 2788
AAACAGAGCGACTCCT-1 1 14 94 3164 7950
AAACAGCTTTCAGAAG-1 1 43 9 6637 2099
AAACAGGGTCTATATT-1 1 47 13 7116 2375
AAACATGGTGAGAGGA-1 1 62 0 8913 1480
然后看看降维聚类等信息(reductions元素)
其中reductions可以是pca,tsne,umap等等,前提是需要对这个对象跑相关的降维代码。比如如果我们跑了代码:
### 我们可以继续对 RNA 表达数据进行降维和聚类,使用与 scRNA-seq 分析相同的工作流程。
brain <- RunPCA(brain, assay = "SCT", verbose = FALSE)
brain <- FindNeighbors(brain, reduction = "pca", dims = 1:30)
brain <- FindClusters(brain, verbose = FALSE)
brain <- RunUMAP(brain, reduction = "pca", dims = 1:30)
就可以看到下面的pca和umap的降维结果啦 :
> brain@reductions
$pca
A dimensional reduction object with key PC_
Number of dimensions: 50
Number of cells: 2696
Projected dimensional reduction calculated: FALSE
Jackstraw run: FALSE
Computed using assay: SCT
$umap
A dimensional reduction object with key umap_
Number of dimensions: 2
Number of cells: 2696
Projected dimensional reduction calculated: FALSE
Jackstraw run: FALSE
Computed using assay: SCT
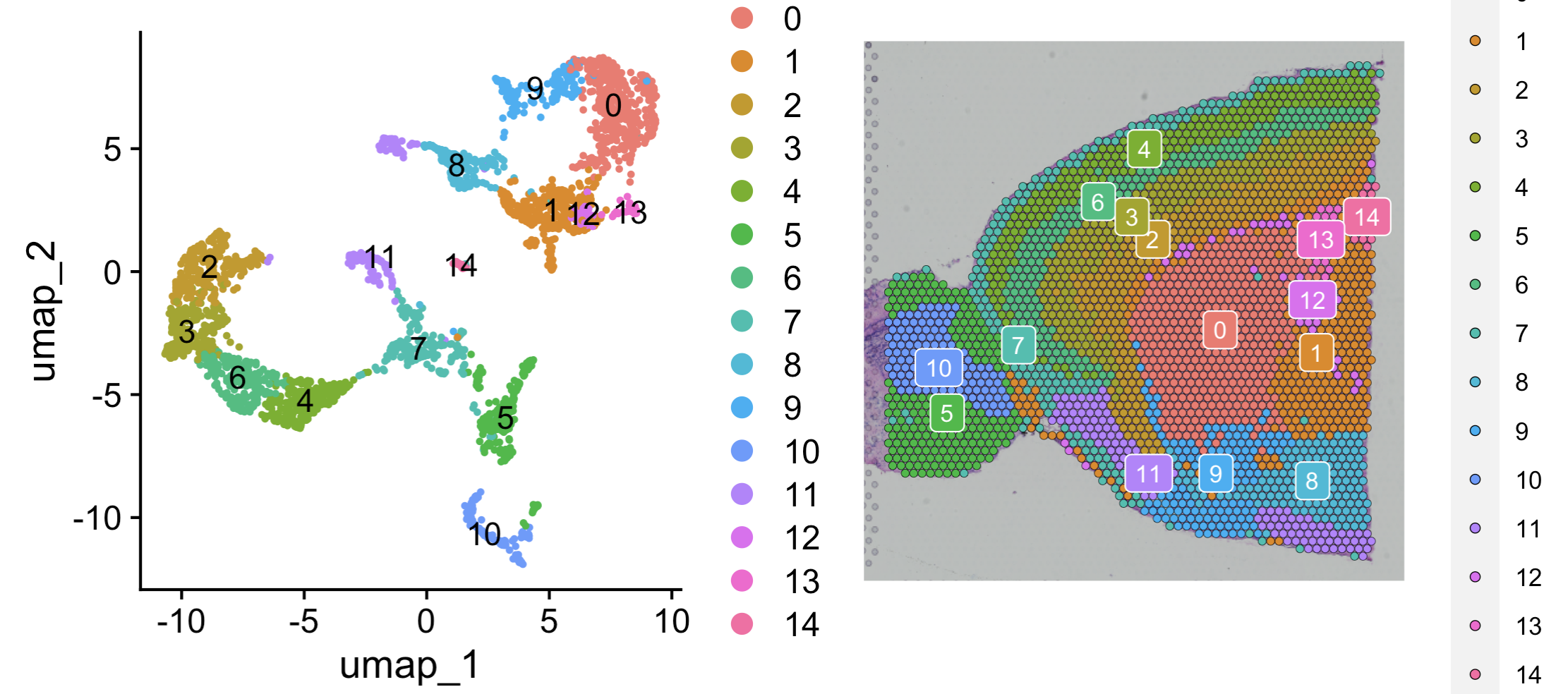
如果有了降维信息的Seurat的空间对象,就可以可视化啦。如下所示的代码:
p1 <- DimPlot(brain, reduction = "umap", label = TRUE)
p1
p2 <- SpatialDimPlot(brain, label = TRUE, label.size = 3)
p2
p1 + p2
效果如下所示(坐标是UMAP的二维坐标散点图,右边是该样品的空间切片坐标):
其它简单信息
比如记录了这个对象在操作过程中(降维聚类分群等等)每个步骤的函数和参数(其实大部分情况下都是默认参数):
> brain@commands
$SCTransform.Spatial
Command: SCTransform(brain, assay = "Spatial", verbose = FALSE)
Time: 2023-12-30 16:36:40.493752
assay : Spatial
new.assay.name : SCT
do.correct.umi : TRUE
ncells : 5000
variable.features.n : 3000
variable.features.rv.th : 1.3
再比如版本信息:
> brain@version
[1] ‘5.0.1’
如果是两个数据对象的合并
代码如下所示:
## 老鼠大脑的这个数据集包含另一个对应于大脑另一半的切片。在这里,我们将其读入并执行相同的初始归一化。
brain2 <- LoadData("stxBrain", type = "posterior1") #直接读取另一半大脑的数据
brain2 <- SCTransform(brain2, assay = "Spatial", verbose = FALSE) #直接用SCTransform代替标准化、找高变与归一化三步
# 为了在同一个 Seurat 对象中处理多个切片,我们提供了该merge函数。
brain.merge <- merge(brain, brain2) #校正后的数据,两个脑切片一起整合
# 然后,这可以对基础 RNA 表达数据进行联合降维和聚类。
DefaultAssay(brain.merge) <- "SCT"
VariableFeatures(brain.merge) <- c(VariableFeatures(brain), VariableFeatures(brain2))
brain.merge <- RunPCA(brain.merge, verbose = FALSE)
brain.merge <- FindNeighbors(brain.merge, dims = 1:30)
brain.merge <- FindClusters(brain.merge, verbose = FALSE)
brain.merge <- RunUMAP(brain.merge, dims = 1:30)
DimPlot(brain.merge, reduction = "umap", group.by = c("ident", "orig.ident"))
SpatialDimPlot(brain.merge)
当然了,绝大部分情况下没有必要合并,因为每个样品自己的空间切片坐标信息是自己的。所以它最后的可视化仍然是每个样品独立,如下所示:

示范数据(Slide-seq技术)
来源于:https://satijalab.org/seurat/articles/spatial_vignette.html#slide-seq
library(SeuratData)
options(timeout=100000)
InstallData("ssHippo")
slide.seq <- LoadData("ssHippo")
slide.seq
因为也是Seurat的空间单细胞对象,所以基本上跟上面介绍的小鼠的大脑的空间切片单细胞转录组同样的数据分析思路。
标准10x技术空间单细胞文件的导入后的对象结构
需要理解Seurat包的一些跟10x技术空间单细胞文件的导入相关的函数,比如:
Read10X Load in data from 10X
Read10X_h5 Read 10X hdf5 file
Read10X_Image Load a 10X Genomics Visium Image
Load10X_Spatial Load a 10x Genomics Visium Spatial Experiment into a 'Seurat' object
这些细节都可以在:https://rdrr.io/github/satijalab/seurat/src/R/preprocessing.R 里面找到。
如果是标准10x技术空间单细胞文件,只需要 Load10X_Spatial 函数即可一步到位,所以就不再赘述啦。如果想搞清楚什么是标准10x技术空间单细胞文件,详见:10x的空间单细胞文件格式详解