最近开始在学习基于R语言的seurat包的单细胞测序数据分析。Jimmy老师给了一个实战分析,在分析过程中逐渐认识seurat包的数据结构。本次推文主要分享一下如何替换seurat对象中的orig.ident为样本名字。
假设我们有一个seurat对象sce.all,默认的每个cell的样本来源信息是存储在sce.all对象中metadata的orig.ident部分(sce.all@meta.data$orig.ident),但是orig.ident中的内容是从1开始的数值,有N个样本,就有N个数值去代表这N个样本。在后续的作图分析中,如果样本信息只是数值,还要对应回原本的样本名去查看,这样并不直观。因此我们有这个需要将seurat对象中的orig.ident替换为真实的样本名字。
批量读入多个10x技术的单细胞转录组样品
首先我们猜想样本读入的顺序应该对应于orig.ident中的1,2,3,4,5…的顺序。我们通过读入后每个样本中的细胞数目与读入前barcodes.tsv.gz的细胞数目来验证猜想。我们先获得样本的路径,然后构建seurat对象(一共8个样本)。最后的部分即为读入后每个样本中的细胞数目。
tmp = list.dirs('outputs/',)
tmp
#tmp=tmp[grepl('filtered_feature_bc_matrix',tmp)]
tmp=tmp[-1]
tmp
basename(tmp)
library(Seurat)
ct = Read10X(tmp)
colnames(ct)
sce.all=CreateSeuratObject(counts = ct ,
min.cells = 5,
min.features = 300)
table(sce.all$orig.ident)
可以看到, 是如下所示的8个样品:
> tmp
[1] "outputs//HRD1"
[2] "outputs//HRD2"
[3] "outputs//HRD3"
[4] "outputs//HRD4"
[5] "outputs//non-HRD1"
[6] "outputs//non-HRD2"
[7] "outputs//non-HRD3"
[8] "outputs//non-HRD4"
> basename(tmp)
[1] "HRD1" "HRD2" "HRD3"
[4] "HRD4" "non-HRD1" "non-HRD2"
[7] "non-HRD3" "non-HRD4"
因为是8个样品同一时间读入了,所以他们的样品名字被抹除了,变成了顺序编号,这样是不合理的。
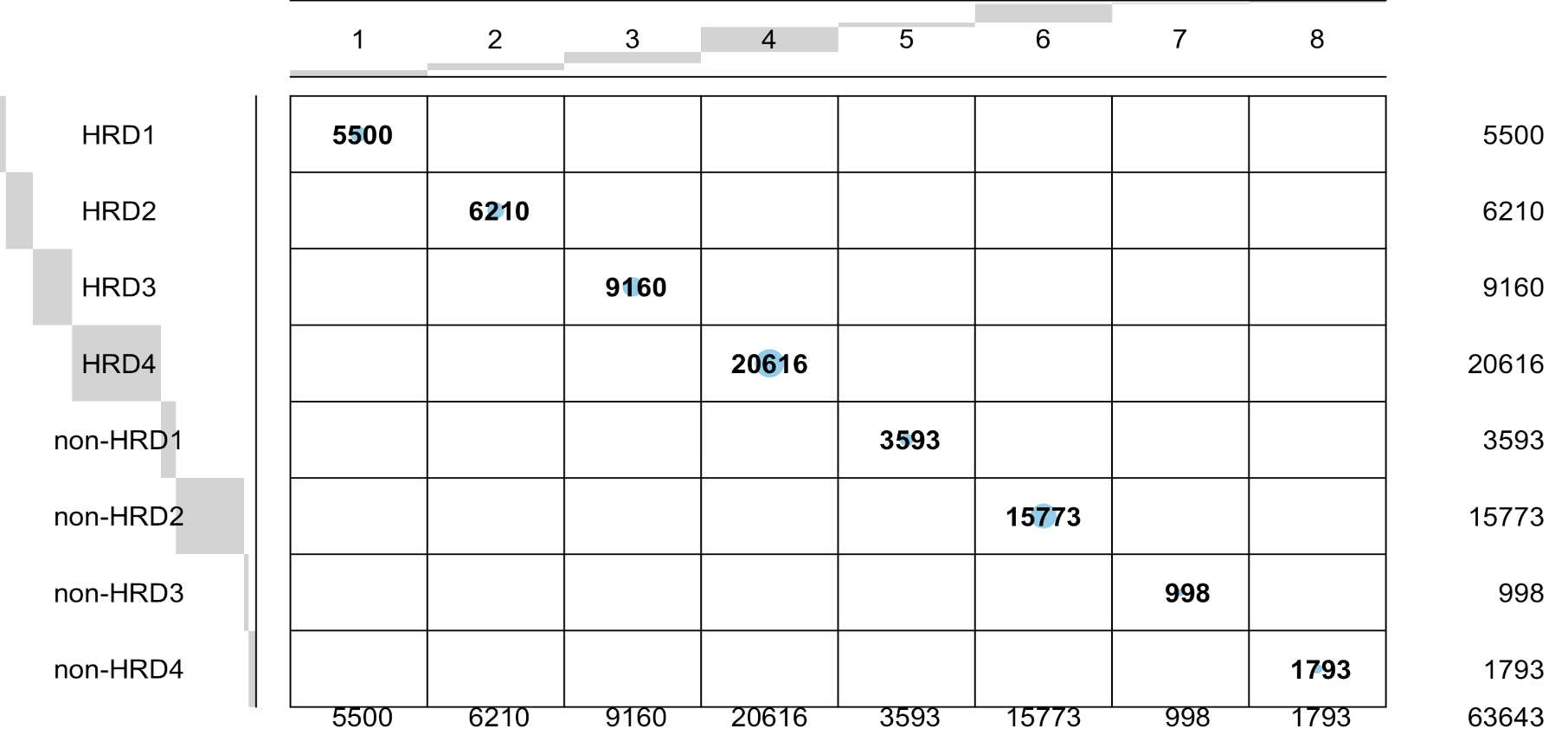
> as.data.frame(table(sce.all$orig.ident))
Var1 Freq
1 1 5500
2 2 6210
3 3 9160
4 4 20616
5 5 3593
6 6 15773
7 7 998
8 8 1793
读入这些标准的10x的单细胞转录组的表达量矩阵文件的代码可以参考基于Seurat的V5版本的系列笔记哦:
检测样品对应关系
现在的问题是,我们的8个样品的具体的样品名字被抹除了,但是我们不清楚样品名字的顺序是否对应着编号数值,所以我们一个个读取看看具体的细胞数量:
lapply(tmp, function(x){
print(x)
print(dim(CreateSeuratObject(counts = Read10X(x) ,
min.cells = 5,
min.features = 300)))
})
结果是:
[1] "outputs//HRD1"
[1] 17864 5500
[1] "outputs//HRD2"
[1] 18063 6210
[1] "outputs//HRD3"
[1] 20717 9160
[1] "outputs//HRD4"
[1] 20949 20616
[1] "outputs//non-HRD1"
[1] 17991 3593
[1] "outputs//non-HRD2"
[1] 19134 15773
[1] "outputs//non-HRD3"
[1] 15798 998
[1] "outputs//non-HRD4"
[1] 16649 1793
可以看到,顺序是一致的,现在就是把样品名字替换即可,需要找到合适的方法和函数:
替换seurat对象中的orig.ident为样本名字
我在bing搜索,(https://www.bing.com/) 输入:rename identiy in seurat,发现 Seurat (version 1.2.1) 是有对应的函数:
rename.ident(object, old.ident.name = NULL, new.ident.name = NULL)
但是我们现在都是Seurat的v5啦,可以去 :https://satijalab.org/seurat/articles/essential_commands.html
看到了全部的相关函数,其实就有RenameIdents函数,简单的方法如下所示:
new.cluster.ids <- basename(tmp)
names(new.cluster.ids) <- levels(sce.all$orig.ident)
pbmc <- RenameIdents(sce.all, new.cluster.ids)
gplots::balloonplot(table(sce.all$orig.ident,Idents(pbmc)))
sce.all$orig.ident = Idents(pbmc)
说明改名是很成功的:
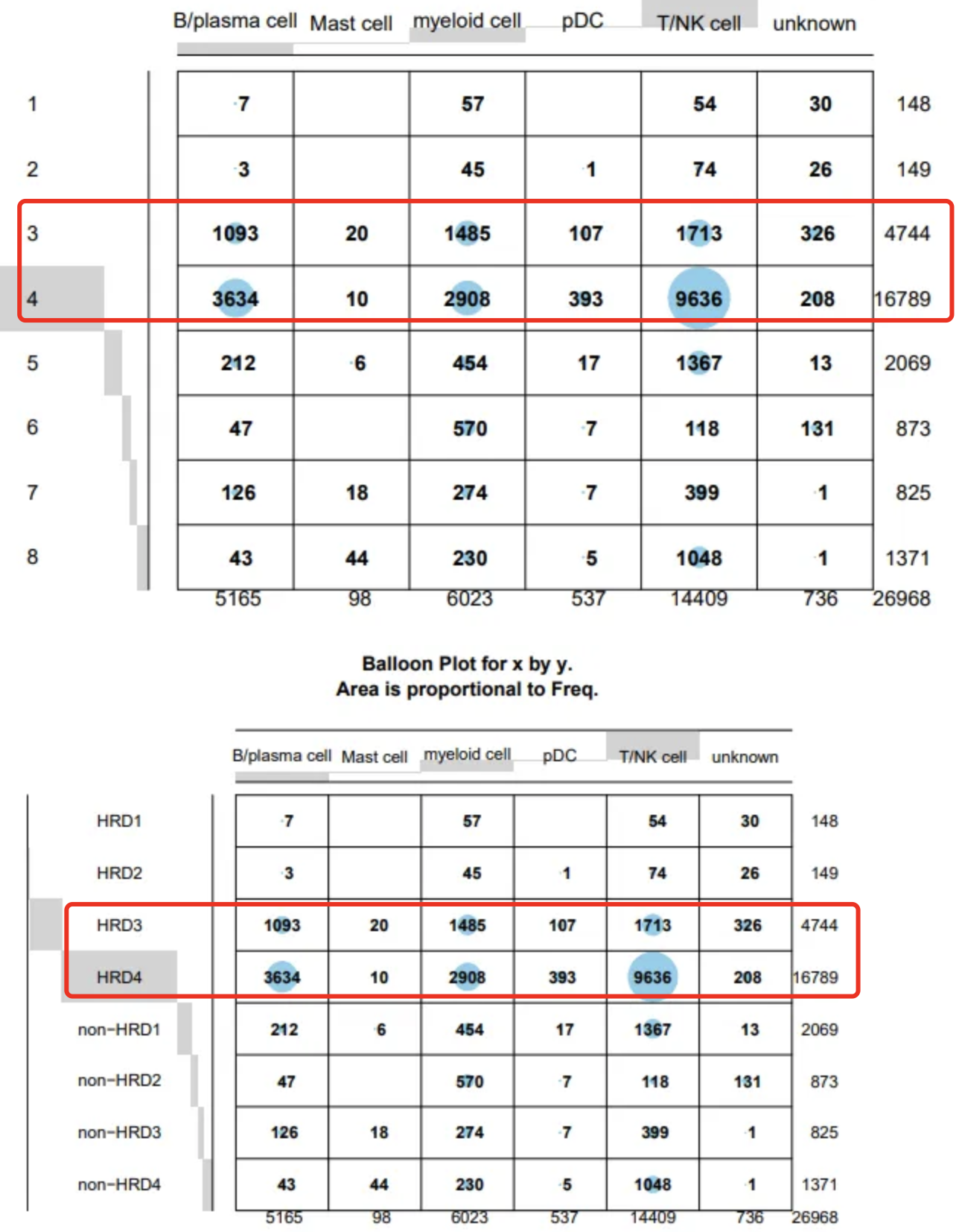
而且我们对上面的对象降维聚类分群后,提取免疫细胞继续细分,可以看到改名与否并不影响任何数据分析结果 :