学员做了一个时间序列转录组测序项目,我们帮忙处理了一下,实验设计如下所示:
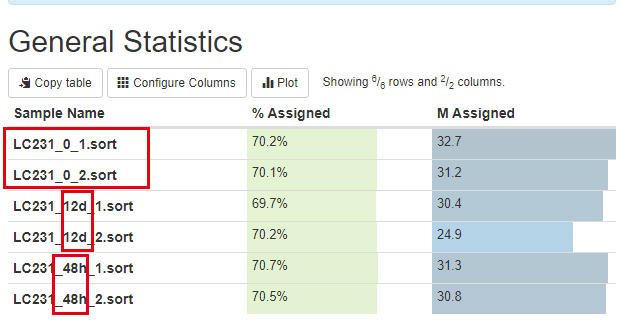
首先是质量控制
可以看到是12d和48h处理的效果不一样,其中48h的处理跟control更接近,而12d的处理很明显影响很大:
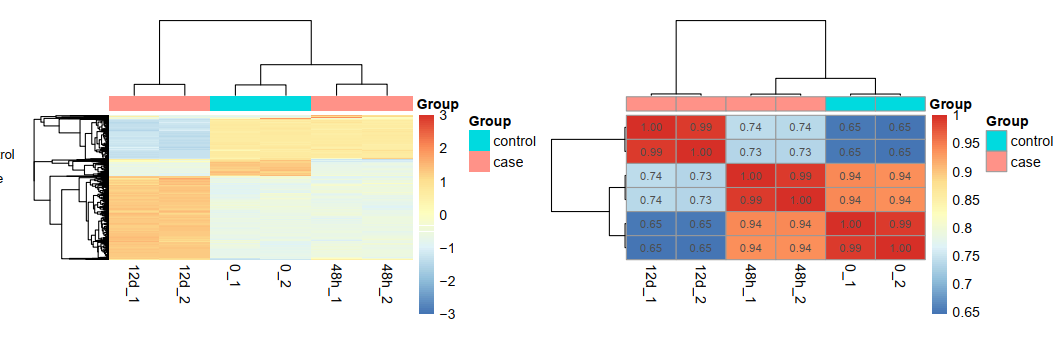
需要多次差异分析
因为是很简单的3分组,0是control组,然后是12d和48h两个不同时间点的处理组,起码可以做如下所示的四次差异分析
- 12d和48h都是treat组,去跟control对比
- 12d,去跟control对比
- 48h,去跟control对比
- 12d和48h对比
每次差异分析都可以出几千个图,非常可怕!就跟单细胞转录组降维聚类分群一样的容易数据集一个标准流程也是出几千个图,但是我们有一个找核心的方法!不同差异分析结果,可以韦恩图,散点图,各种。。。
推荐时间序列分析(mfuzz)
时序分析可以找核心变化,其实就是针对基因的聚类分群而已,wgcna算法也是如此!
实战演练
一般来说,差异分析流程适用于两两比较, 但实际科研中的实验设计会比较复杂,比如:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE116439
里面的其中一个细胞系的一种药物的不同浓度不同时间段处理数据如下:
GSM3232610 A498_cisplatin_0nM_24h
GSM3232611 A498_cisplatin_0nM_2h
GSM3232612 A498_cisplatin_0nM_6h
GSM3232613 A498_cisplatin_15000nM_24h
GSM3232614 A498_cisplatin_15000nM_2h
GSM3232615 A498_cisplatin_15000nM_6h
GSM3232616 A498_cisplatin_3000nM_24h
GSM3232617 A498_cisplatin_3000nM_2h
GSM3232618 A498_cisplatin_3000nM_6h
这个项目本身很复杂,Drug-induced change in gene expression across NCI-60 cell lines after exposure to 15 anticancer agents for 2, 6 and 24h (cisplatin)
但是,我们只需要针对里面的一个细胞系的一种药物的不同浓度不同时间段处理做一个简单的时间序列转录组多次差异分析以及时序分析!
总体上来说,数据分析思路,跟前面的时间序列转录组测序多次差异分析以及时序分析一样的,前面的是转录组测序,现在举例的这个GSE116439是转录组的表达量芯片!我们在GEO的官网下载:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE116439 如下所示的文件:
2.4M Jun 29 2018 GSM3232610_A498_cisplatin_0nM_24h_U133A.CEL.gz
2.4M Jun 29 2018 GSM3232611_A498_cisplatin_0nM_2h_U133A.CEL.gz
2.5M Jun 29 2018 GSM3232612_A498_cisplatin_0nM_6h_U133A.CEL.gz
2.5M Jun 29 2018 GSM3232613_A498_cisplatin_15000nM_24h_U133A.CEL.gz
2.4M Jun 29 2018 GSM3232614_A498_cisplatin_15000nM_2h_U133A.CEL.gz
2.4M Jun 29 2018 GSM3232615_A498_cisplatin_15000nM_6h_U133A.CEL.gz
2.4M Jun 29 2018 GSM3232616_A498_cisplatin_3000nM_24h_U133A.CEL.gz
2.4M Jun 29 2018 GSM3232617_A498_cisplatin_3000nM_2h_U133A.CEL.gz
2.4M Jun 29 2018 GSM3232618_A498_cisplatin_3000nM_6h_U133A.CEL.gz
处理表达量芯片的cel文件成为表达量矩阵
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
getOption('timeout')
options(timeout=10000)
library(oligo)
library(ggplot2)
# BiocManager::install('pd.ht.hg.u133a')
celFiles <- list.celfiles('GSE116439_RAW/',listGzipped = T,
full.name=TRUE)
celFiles
exon_data <- oligo::read.celfiles( celFiles )
dim(exprs(exon_data))
exprs(exon_data)[1:4,1:4]
### 标准化(一步完成背景校正、分位数校正标准化和RMA (robust multichip average) 表达整合)
exon_data_rma <- oligo::rma(exon_data )
exp_probe <- Biobase::exprs(exon_data_rma)
exp_probe[1:4,1:4]
boxplot(exp_probe[,1:4],las=2)
dat = exp_probe
boxplot(dat[,1:4],las=2)
library(limma)
#dat=normalizeBetweenArrays(dat)
boxplot(dat[,1:4],las=2)
library(stringr)
#pd=pData(a)
pd = as.data.frame(str_split(colnames(dat),'_',simplify = T)[,c(4,5)])
#通过查看说明书知道取对象a里的临床信息用pData
## 挑选一些感兴趣的临床表型。
library(stringr)
# pd=pd[1:43,]
# dat=dat[,1:43]
phe=pd
# 这里一定要人工干预,每个数据集项目的分组不一样
# 是主观判断,自己选择
#group_list=ifelse(grepl('Healthy',phe$title),'healthy','patient')
group_list=phe$V2
table(group_list)
#tmp = pd[group_list=='healthy',]
# 996 samples analyzed of which 72 are Healthy controls and 924 are SLE;
dat[1:4,1:4]
dim(dat)
library(AnnoProbe)
ids=idmap('GPL571','soft')
head(ids)
ids=ids[ids$symbol != '',]
dat=dat[rownames(dat) %in% ids$ID,]
ids=ids[match(rownames(dat),ids$ID),]
head(ids)
colnames(ids)=c('probe_id','symbol')
ids$probe_id=as.character(ids$probe_id)
rownames(dat)=ids$probe_id
dat[1:4,1:4]
ids=ids[ids$probe_id %in% rownames(dat),]
dat[1:4,1:4]
dat=dat[ids$probe_id,]
ids$median=apply(dat,1,median) #ids新建median这一列,列名为median,同时对dat这个矩阵按行操作,取每一行的中位数,将结果给到median这一列的每一行
ids=ids[order(ids$symbol,ids$median,decreasing = T),]#对ids$symbol按照ids$median中位数从大到小排列的顺序排序,将对应的行赋值为一个新的ids
ids=ids[!duplicated(ids$symbol),]#将symbol这一列取取出重复项,'!'为否,即取出不重复的项,去除重复的gene ,保留每个基因最大表达量结果s
dat=dat[ids$probe_id,] #新的ids取出probe_id这一列,将dat按照取出的这一列中的每一行组成一个新的dat
rownames(dat)=ids$symbol#把ids的symbol这一列中的每一行给dat作为dat的行名
dat[1:4,1:4] #保留每个基因ID第一次出现的信息
dat['ACTB',]
dat['GAPDH',]
save(dat,group_list,phe,file = 'step1-output.Rdata')
source('step2-check.R')
# 被迫忽略这个浓度信息
质量控制发现3个时间点确实是差异很大
质量控制的代码在 source(‘step2-check.R’) 里面,可以看到:
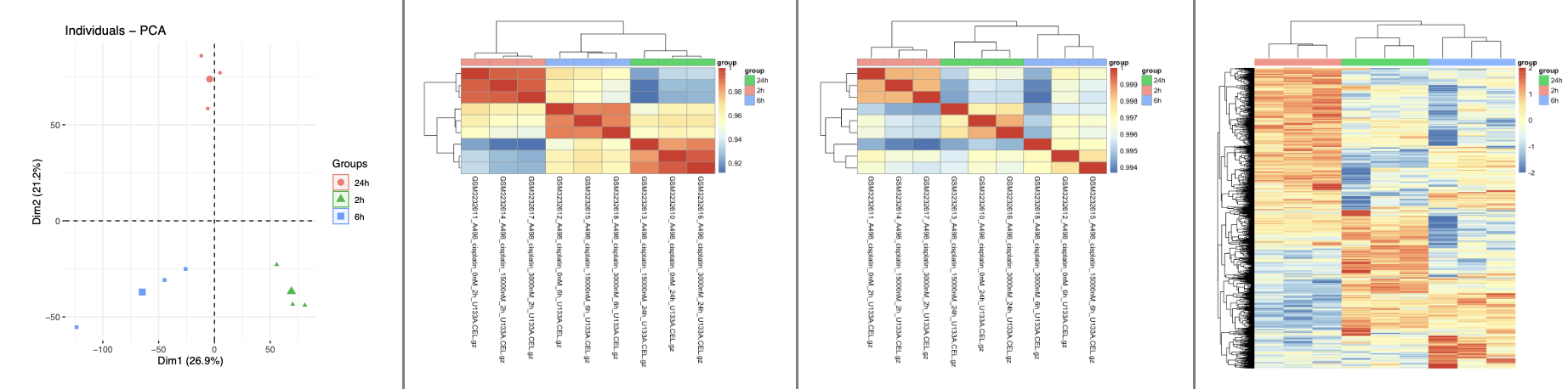
两两组合的差异分析
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
library(clusterProfiler)
library(stringr)
library(limma)
run <- function(){
getwd()
source('../functions/run_check.R')
run_check(dat, group_list, target_gene='GAPDH', pro=gse_number,path='./')
source('../functions/run_DEG.R')
group_list
table(group_list)
deg=run_DEG(dat, group_list, pro=gse_number,path='./',
target_gene=c('GAPDH'))
head(deg)
source('../functions/run_ORA_KEGG.R')
# Reading KEGG annotation online:
run_ORA_KEGG(deg, path='./')
source('../functions/run_ORA_GO.R')
run_ORA_GO(deg, path='./')
load(file = 'anno_DEG.Rdata')
source('../functions/run_GSEA_KEGG.R')
run_GSEA_KEGG(DEG, path='./')
}
load(file = 'step1-output.Rdata')
table(group_list)
# 每次都要检测数据
dat[1:4,1:4]
raw_dat = dat
raw_gp = group_list
pro='6h-vs-2h'
dir.create(pro)
setwd(pro)
table(raw_gp)
kp = raw_gp != '24h'
table(kp)
dat = raw_dat[,kp]
group_list = raw_gp[kp]
gse_number = pro
save( gse_number,
dat,group_list,
file = 'step1-output.Rdata')
load(file = 'step1-output.Rdata')
run()
setwd('../')
getwd()
pro='24h-vs-2h'
dir.create(pro)
setwd(pro)
table(raw_gp)
kp = raw_gp != '6h'
table(kp)
dat = raw_dat[,kp]
group_list = raw_gp[kp]
gse_number = pro
save( gse_number,
dat,group_list,
file = 'step1-output.Rdata')
load(file = 'step1-output.Rdata')
run()
setwd('../')
getwd()
pro='24h-vs-6h'
dir.create(pro)
setwd(pro)
table(raw_gp)
kp = raw_gp != '2h'
table(kp)
dat = raw_dat[,kp]
group_list = raw_gp[kp]
gse_number = pro
save( gse_number,
dat,group_list,
file = 'step1-output.Rdata')
load(file = 'step1-output.Rdata')
run()
setwd('../')
getwd()
只需要从原始的表达量矩阵里面任意提取需要的二分组表达量矩阵即可批量做差异分析富集分析,而且每次分析都有大量的图表结果, 如下学习:
我们当然是可以具体去看每次差异分析的上下调基因和通路,但是它不够聚焦,尤其是时间点变多的时候,差异分析的两两分组组合起来可以是几十次分析, 图表就成千上万了!
核心基因和通路分析
那就是mfuzz这样的时序分析啦,说简单一点就是把基因进行分组而已,代码也很简单,分组可以自定义需要多少组,我这里随意举例是9个分组,如下所示:
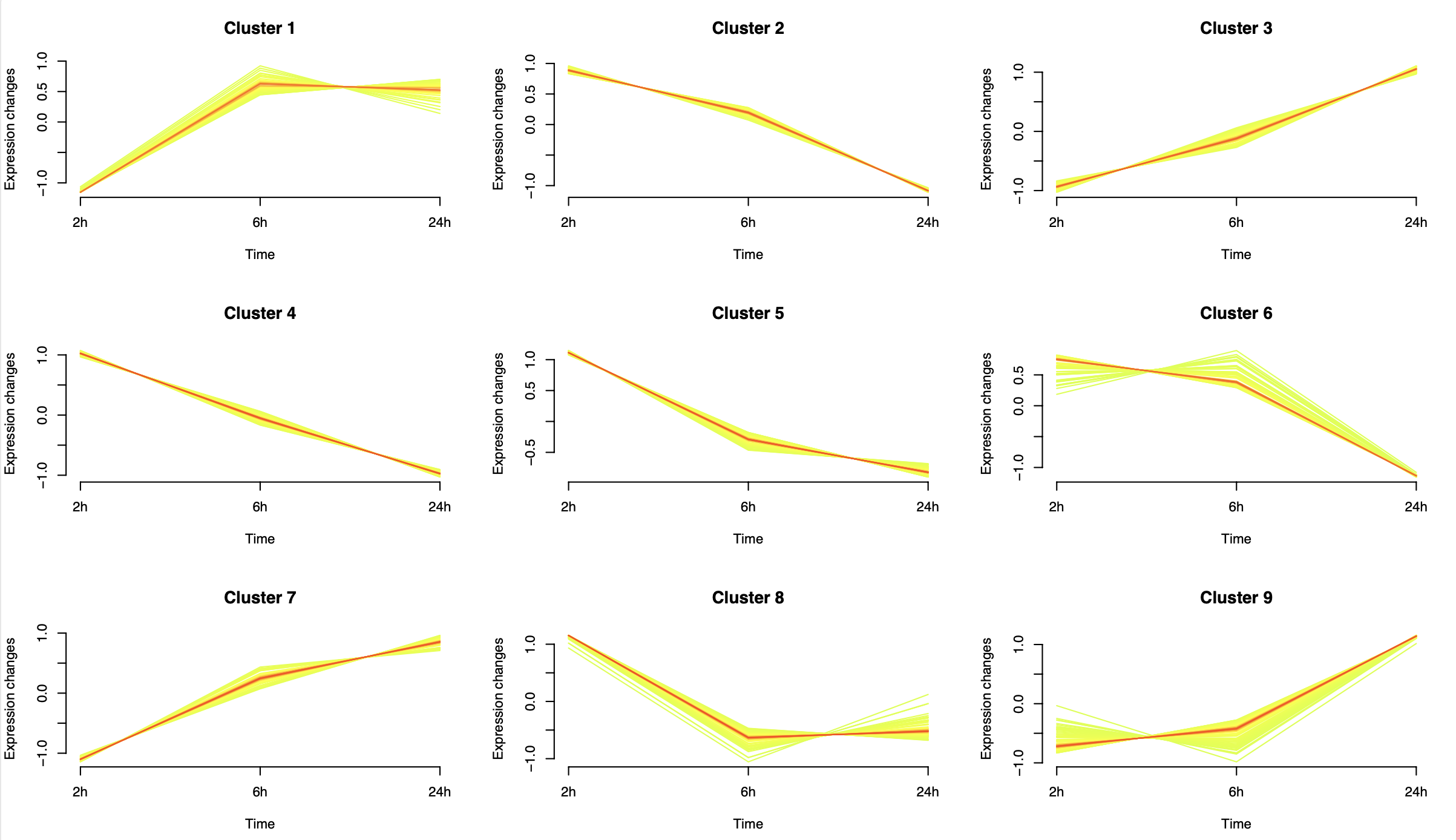
因为最开始我们就选择了3次差异分析的统计学显著的基因,就不到600个,所以拆分成为了9个分组后每个分组里面的基因数量并不多
> table(group_g$group)
c1 c2 c3 c4 c5 c6 c7 c8 c9
48 38 54 57 57 49 53 51 77
简单的统计学功能注释如下所示:
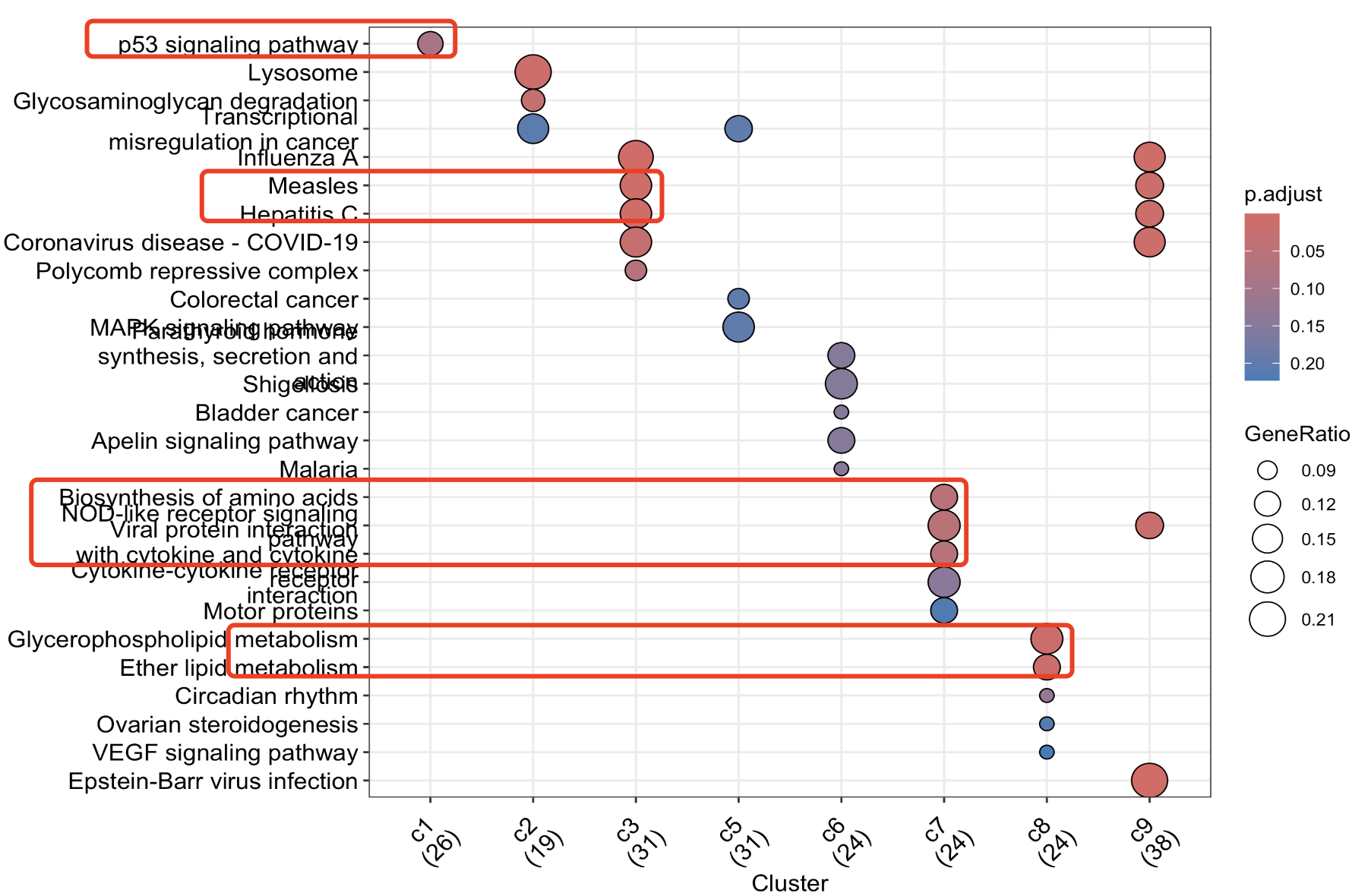
确实是代码有点多
我简单的看了看,仅仅是每个函数就都是一百多行啦,是我过去半年的“大杂烩”:
wc -l functions/*
84 functions/enrich.R
137 functions/run_DEG.R
122 functions/run_DEG_by_DESeq2.R
75 functions/run_GSEA_KEGG.R
64 functions/run_ORA_GO.R
93 functions/run_ORA_KEGG.R
116 functions/run_check.R
122 functions/run_gseKEGG.R
有偿获取上面的全部代码
从表达量芯片的cel文件开始处理得到表达量矩阵,提取3个分组信息,两两组合差异分析3次,然后mfuzz分析对差异基因进行分组,分组后进行生物学功能数据库注释,如果是从零开始可以根据我的代码慢慢学习,肯定是可以跑通!

如果上面的群聊二维码满员了,大家可以扫描下面的二维码添加我的秘书(每天晚上十点钟统一给大家发放代码并且拉群)
