前些天我在 生物学功能注释三板斧,提到了简单的超几何分布检验,复杂一点可以是gsea和gsva,更复杂一点的可以是DoRothEA和PROGENy类似的打分。
其中 GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库是两个常用的生物学功能注释数据库,科学家通常是使用来超几何分布检验这个统计学算法做富集分析,即通过比较实际观察到的基因集合(几十个或者几百个)中特定功能或通路的基因数量与随机期望的数量来判断其是否富集。
但是GO数据库 注释通常包括三个方面的信息:分子功能(Molecular Function)、细胞组分(Cellular Component)和生物过程(Biological Process)。而且它有一个特点是有向无环图(DAG),用一个简单的比喻来理解:
- 树状结构: 想象一棵大树,树干是GO的根节点,而树枝和叶子是各种不同的基因功能术语。
- 节点和边: 在这个家谱树上,每一个节点代表一个特定的基因功能术语,例如“细胞分裂”、“代谢过程”等。而边(连接节点的线)表示这些术语之间的关系。
- 层次关系: 树的不同层次代表了基因功能的层次关系。树干是一些更通用、更广泛的术语,而分支和叶子是更具体和详细的术语。
- 无环: DAG是有向无环图,这意味着在图中你不会发现任何形成循环的路径。从根节点到任何一个节点都是沿着一个单一的方向,没有回头。
普通的GO数据库注释
简单的转录组差异分析
首先我们获取airway这个包里面的airway表达量矩阵,它有8个样品,分成了两组,我们命名为’control’,’case’ ,如下所示代码:
library(data.table)
library(airway,quietly = T)
data(airway)
mat <- assay(airway)
keep_feature <- rowSums (mat > 1) > 1
ensembl_matrix <- mat[keep_feature, ]
library(AnnoProbe)
ids=annoGene(rownames(ensembl_matrix),'ENSEMBL','human')
ids=ids[!duplicated(ids$SYMBOL),]
ids=ids[!duplicated(ids$ENSEMBL),]
symbol_matrix= ensembl_matrix[match(ids$ENSEMBL,rownames(ensembl_matrix)),]
rownames(symbol_matrix) = ids$SYMBOL
group_list = as.character(airway@colData$dex);group_list
group_list=ifelse(group_list=='untrt','control','case' )
group_list = factor(group_list,levels = c('control','case' ))
然后针对上面的airway这个包里面的airway表达量矩阵,是一个简单的转录组测序后的count矩阵,可以走DESeq2进行转录组差异分析,代码如下所示:
(colData <- data.frame(row.names=colnames(symbol_matrix),
group_list=group_list) )
dds <- DESeqDataSetFromMatrix(countData = symbol_matrix,
colData = colData,
design = ~ group_list)
dds <- DESeq(dds)
res <- results(dds,
contrast=c("group_list",
levels(group_list)[2],
levels(group_list)[1]))
resOrdered <- res[order(res$padj),]
DEG =as.data.frame(resOrdered)
DEG_deseq2 = na.omit(DEG)
差异分析的结果矩阵比较简单:
> head(DEG_deseq2)
baseMean log2FoldChange lfcSE stat pvalue padj
SPARCL1 997.2841 4.601648 0.21173761 21.73278 1.005191e-104 1.751345e-100
STOM 11193.6206 1.450654 0.08458760 17.14973 6.314974e-66 5.501289e-62
PER1 776.4988 3.183045 0.20133158 15.80996 2.656240e-56 1.542656e-52
PHC2 2738.0273 1.386358 0.09161873 15.13182 9.988847e-52 4.350892e-48
MT2A 3656.0130 2.202678 0.14718400 14.96547 1.234488e-50 4.301697e-47
DUSP1 3408.8006 2.948154 0.20174509 14.61326 2.311720e-48 6.712850e-45
上面的这些列的简要介绍是:
- baseMean:
- 表达水平的均值,表示基因在不同条件或组合中的平均表达水平。
- log2FoldChange:
- 表达差异的对数倍数变化,表示基因在两个条件或组合之间的表达变化倍数的对数。
- lfcSE:
- 对数倍数变化的标准差,用于衡量差异的稳定性。
- stat:
- 统计检验的值,通常是log2FoldChange除以lfcSE,用于判断基因表达的变化是否显著。
- pvalue:
- 统计检验的p值,表示在零假设下观察到的差异或效应发生的概率。
- padj:
- 经过多重检验校正后的p值,用于控制多重检验的误差,提高差异的可靠性。
然后使用最流行的clusterProfiler进行GO数据库的注释
前面提到了,GO数据库 注释通常包括三个方面的信息:分子功能(Molecular Function)、细胞组分(Cellular Component)和生物过程(Biological Process)。我们这里先拿生物过程(Biological Process)举例子吧.
前面的DESeq2进行转录组差异分析后的表格里面有两万多个基因,但是我们根据里面的log2FoldChange对基因排序后取 log2FoldChange 最大的1000个基因来使用超几何分布检验这个统计学算法做富集分析,代码如下所示:
DEG_deseq2=DEG_deseq2[order(DEG_deseq2$log2FoldChange),]
up_top_genes = AnnotationDbi::select(org.Hs.eg.db,keys=tail(rownames(DEG_deseq2),1000) ,
columns="ENTREZID",keytype = "SYMBOL")[,2]
library(clusterProfiler)
library(org.Hs.eg.db)
go_BP_hyper <- enrichGO(
gene = up_top_genes,
keyType = "ENTREZID",
OrgDb = org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05,
readable = TRUE)
head(go_BP_hyper@result)
可以看到这个结果表格非常的丰富:

在使用clusterProfiler进行GO(Gene Ontology)数据库的富集分析时,得到的结果表格通常包含以下列:
- ID:
- 介绍: GO Term的唯一标识符,用于标识富集的功能术语。
- Description:
- 介绍: GO Term的文本描述,说明富集功能的意义。
- GeneRatio:
- 介绍: 在用户提供的基因集中与某GO Term相关的基因占比。
- BgRatio:
- 介绍: 在整个背景基因集中与某GO Term相关的基因占比。
- pvalue:
- 介绍: 富集分析的p值,表示GO Term富集的显著性。
- p.adjust (或FDR):
- 介绍: 校正后的p值,用于控制多重检验误差。
- qvalue:
- 介绍: 与p.adjust相似,也是校正后的p值。
- geneID:
- 介绍: 在GO Term下具有显著富集的基因的标识符,如果前面有 readable = TRUE的参数,那么这个时候就是基因的symbol,如果没有那就是ENTREZID
- Count:
- 介绍: 在用户提供的基因集中与GO Term相关的基因数量。
那么我们通常是会如何可视化这个最流行的clusterProfiler进行GO数据库的注释:
barplot(go_BP_hyper)
平平无奇的条形图,可以看到最显著的一些通路的名字以及其对应的富集信息:
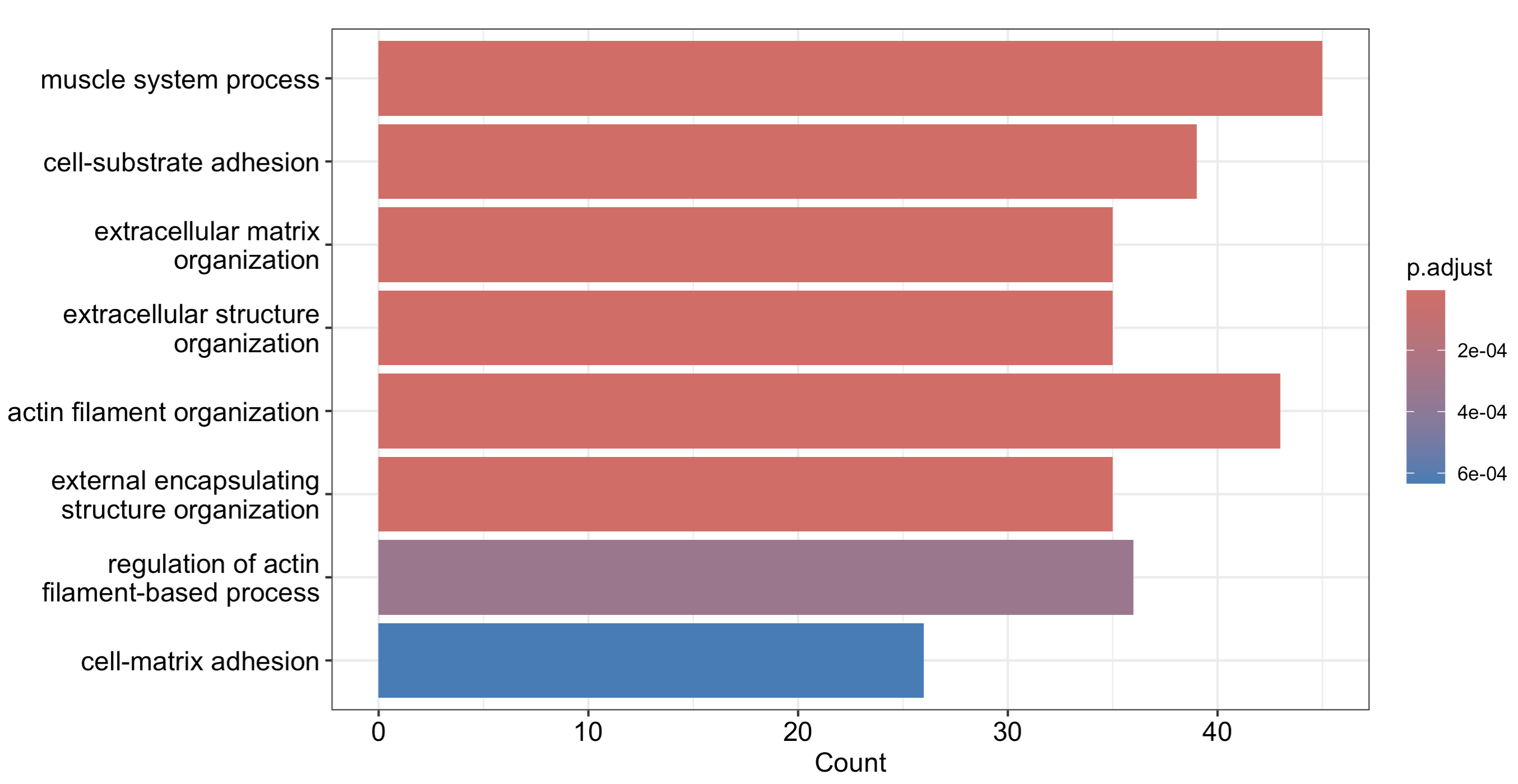
很明显的可以看到上面的通路是有层级关系的,就是前面提到的有向无环图(DAG),而topGO的高级可视化就是帮助我们展示这个有向无环图(DAG)的。
topGO的高级可视化
代码确实是非常简单啦,如下所示:
library(topGO)
goplot(go_BP_hyper,showCategory = 8)
可以看到的是好几个通路的关系 :
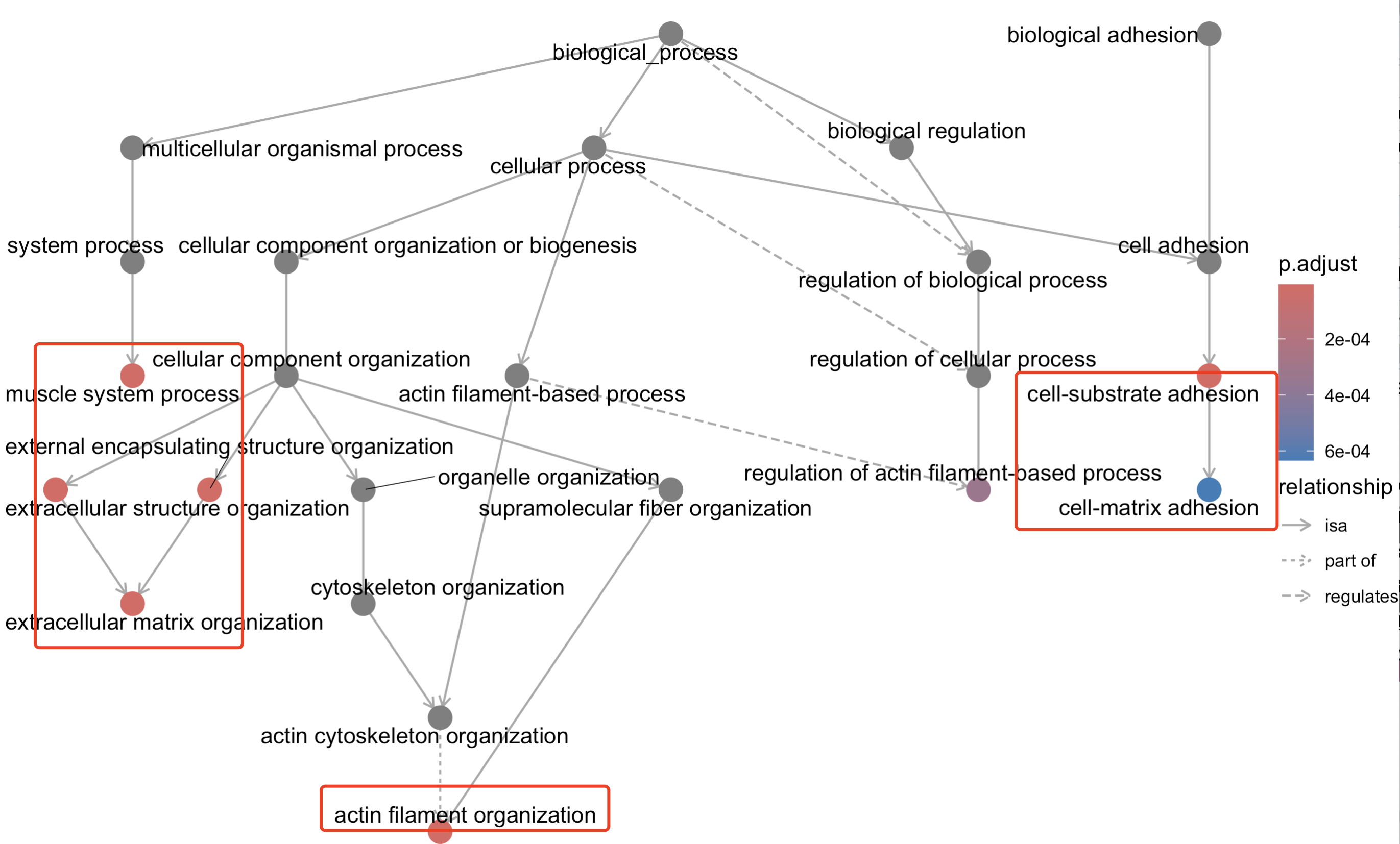
当然了,topGO 是一个用于生物学的Gene Ontology(GO)富集分析的R包,不仅仅是可以展示上面提到的有向无环图(DAG),它还具有以下可视化方面的优点:
- 图形化展示:
topGO提供了直观的图形化展示,例如饼图、条形图等,有助于用户更清晰地理解富集结果。
- GO Term网络图:
- 可以生成GO Term之间的关系网络图,帮助用户理解GO Term的层次结构和相互关系。
- 直观的树状图:
topGO可以生成GO Term的树状图,以树状结构直观地展示GO Term之间的层次关系。
- 用户定制化:
- 允许用户对生成的图形进行定制,包括颜色、标签等,以满足用户个性化的可视化需求。
- 交互式可视化:
- 提供了交互式的可视化功能,用户可以通过交互式控件进行放大、缩小、导出等操作,增强用户体验。
- 基因与GO Term的关联图:
- 可以绘制基因与GO Term之间的关系图,帮助用户了解哪些基因与哪些功能术语关联紧密。
- 支持多种可视化输出:
topGO支持生成多种格式的图形输出,如PDF、PNG等,方便用户在出版物或报告中使用。
- 丰富的图例和注释:
- 生成的图形中包含丰富的图例和注释,使用户更容易理解和解释富集分析的结果。
这些可视化优点使得 topGO 成为生物学研究中进行GO富集分析时一个强大的工具,通过直观的图形展示,帮助研究人员更好地理解基因集与功能术语之间的关系。更多技巧当然是推荐看它的官网啦:
另外,其实上面的富集分析结果是可以精简的, 因为生物过程(Biological Process)有很多冗余,如下所示的代码:
go_BP_simp <- simplify(
go_BP_hyper,
cutoff=0.5,
by="p.adjust",
select_fun=min
)
head(go_BP_simp@result)
dim(go_BP_simp@result)
barplot(go_BP_simp)
goplot(go_BP_simp,showCategory = 8)
另外,类似的增强GO数据库注释结果的可视化的包也非常多, 比如GOplot,可以把结果画出八卦图的样子,同样的也是推荐看官方文档代码啦:
library(ggplot2)
library(GOplot)
data(EC)
head(EC$david)
head(EC$genelist)
circ <- circle_dat(EC$david, EC$genelist)
GOBar(subset(circ, category == 'BP'))
GOBar(circ, display = 'multiple')
# 加标题上色
GOBar(circ, display = 'multiple', title = 'Z-score coloured barplot',
zsc.col = c('yellow', 'black', 'cyan'))
# 分区
GOBubble(circ, labels = 3)
# 加标题, 改变环的颜色, f分区,
GOBubble(circ, title = 'Bubble plot', colour = c('orange', 'darkred', 'gold'), display = 'multiple', labels = 3)
# Colour the background according to the category
GOBubble(circ, title = 'Bubble plot with background colour', display = 'multiple', bg.col = T, labels = 3)
# Reduce redundant terms with a gene overlap >= 0.75...
reduced_circ <- reduce_overlap(circ, overlap = 0.75)
# ...and plot it
GOBubble(reduced_circ, labels = 2.8)
#Generate a circular visualization of the results of gene- annotation enrichment analysis
GOCircle(circ)
# Generate a circular visualization of selected terms
IDs <- c('GO:0007507', 'GO:0001568', 'GO:0001944', 'GO:0048729', 'GO:0048514', 'GO:0005886', 'GO:0008092', 'GO:0008047')
GOCircle(circ, nsub = IDs)
# Generate a circular visualization for 10 terms
GOCircle(circ, nsub = 10)
如下所示的效果:
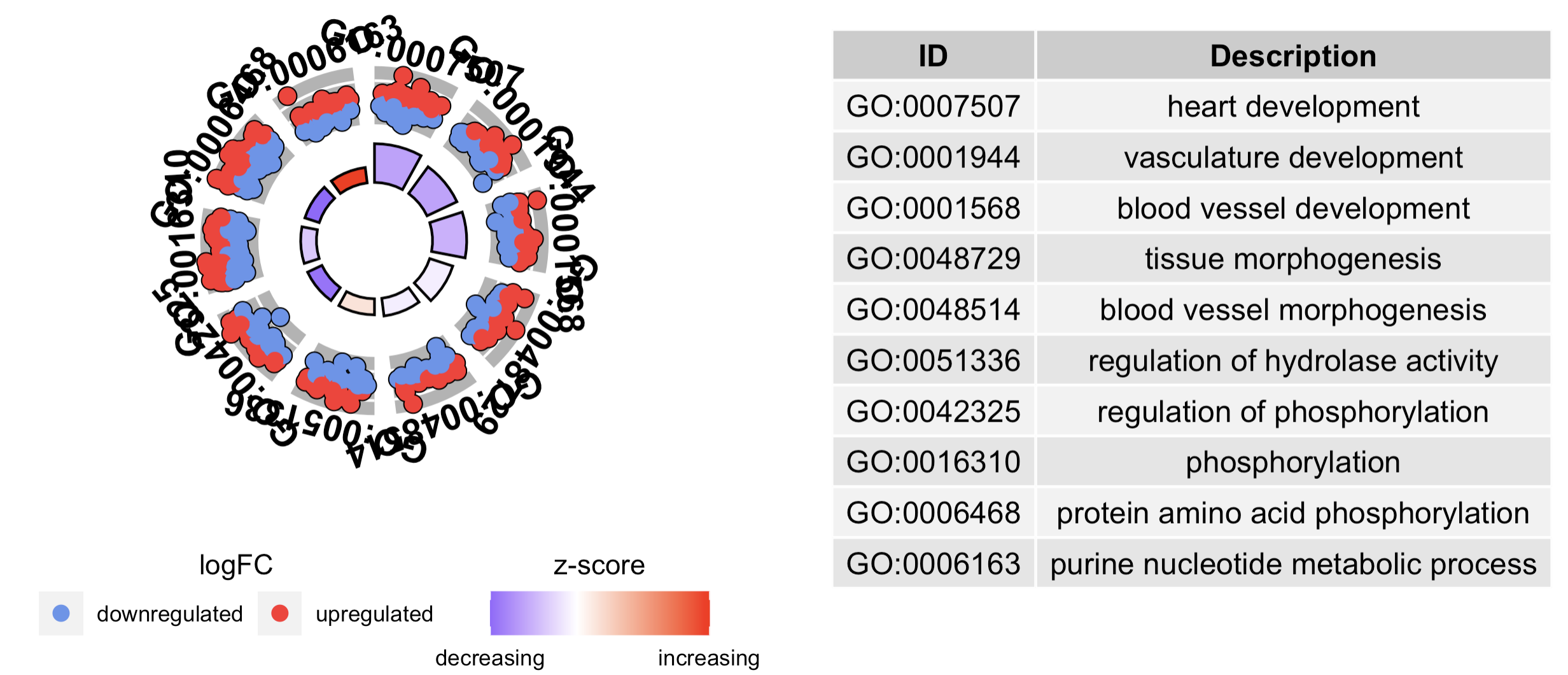
上面的是超几何分布检验的结果的可视化的丰富,其实如果是gsea也有对应的包,后面我们会介绍使用aPEAR来增强clusterProfiler的GSEA分析结果。