前些天在《生信技能树》的微信视频号做了一个肿瘤单细胞转录组的数据分析直播,文章是:《Delineating the dynamic evolution from preneoplasia to invasive lung adenocarcinoma by integrating single-cell RNA sequencing and spatial transcriptomics》详见:换一个分析策略会导致文章的全部论点都得推倒重来吗。
其中第一层次降维聚类分群后给出来了合理的生物学命名,详见:肿瘤单细胞转录组的第一层次降维聚类分群, 整体来说这个复现的代码在百度云分享给大家:链接:https://pan.baidu.com/s/1niFqyAiUU3yXK1W26b8RvQ?pwd=nbmj
如下所示:
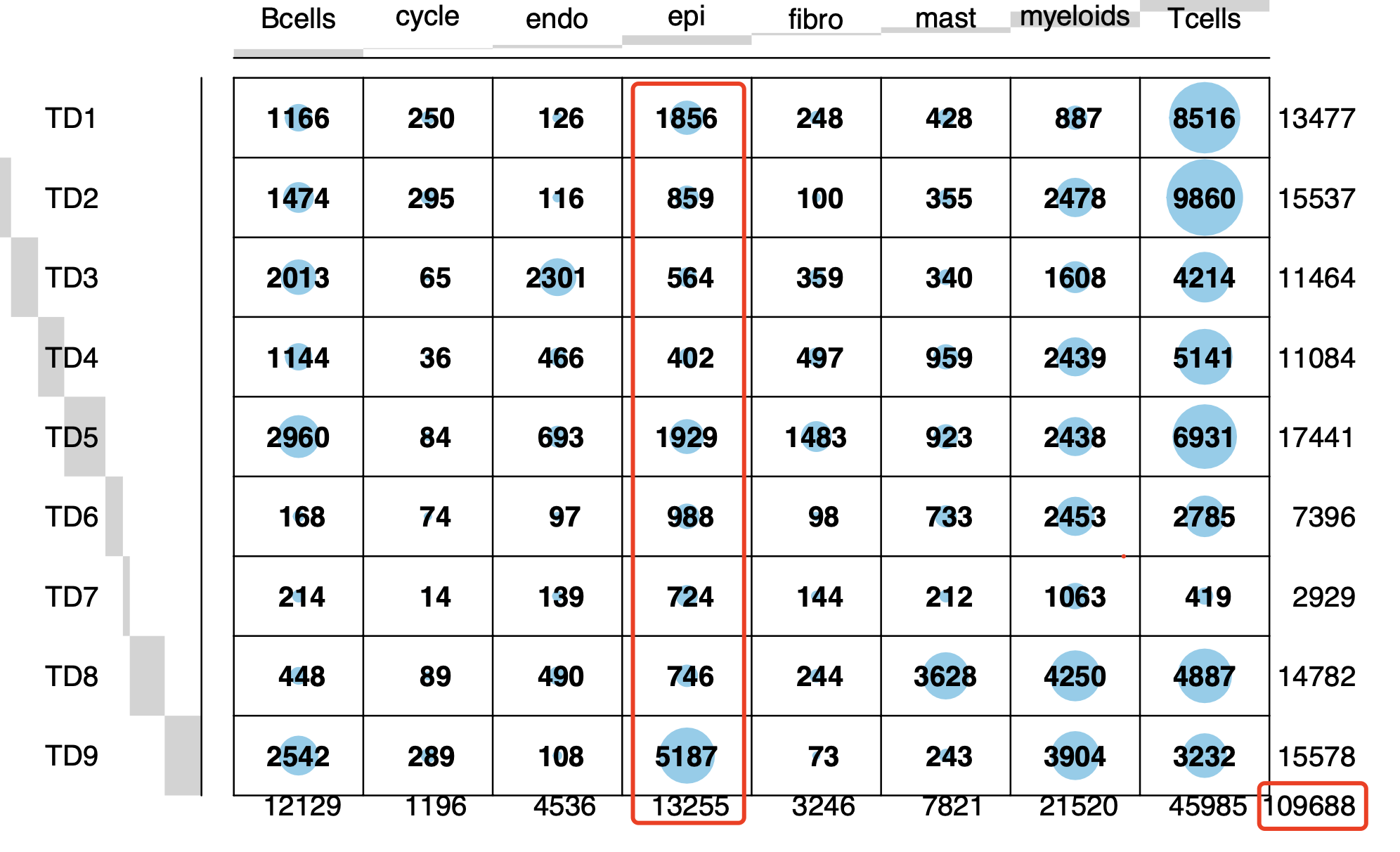
然后,因为文章是针对里面的上皮细胞进行细致的挖掘,所以我们也是如此的演示一下哈:
提取感兴趣亚群子集
值得注意是我们依赖于这个V4的版本的Seurat流程做出来了大量的公共数据集的单细胞转录组降维聚类分群流程,100多个公共单细胞数据集全部的处理,链接:https://pan.baidu.com/s/1MzfqW07P9ZqEA_URQ6rLbA?pwd=3heo
sce.all.int = readRDS('../../2-harmony/sce.all_int.rds')
colnames(sce.all.int@meta.data)
table(sce.all.int$RNA_snn_res.0.8)
load('../../phe.Rdata')
sce.all.int@meta.data =phe
table(sce.all.int$celltype)
# 取子集
sce.all=sce.all.int[,sce.all.int$celltype=='epi']
sce.all=CreateSeuratObject(
counts = sce.all@assays$RNA@counts,
meta.data = sce.all@meta.data
)
as.data.frame(sce.all@assays$RNA@counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all@meta.data$orig.ident)
提取的子集通常是我们会把它重新制作成为了一个几乎是空白的Seurat对象。
继续降维聚类分群和命名
是否整合取决于你的研究目标,如果你想研究的是肿瘤内部异质性(ITH),一般来说是针对每个个体进行NMF等找关键基因列表,然后汇总。
但是如果想研究的是队列,那么就可以把这些上皮细胞harmony整合,然后区分正常细胞和恶性细胞后,看不同上皮细胞亚群的临床意义,比如这个文章就是如此。
继续降维聚类分群和命名的复现的代码在百度云分享给大家:链接:https://pan.baidu.com/s/1niFqyAiUU3yXK1W26b8RvQ?pwd=nbmj
肿瘤单细胞转录组独特的拷贝数分析
需要把这些上皮细胞harmony整合,然后区分正常细胞和恶性细胞后,这个时候跑的是inferCNV流程,详见:肿瘤单细胞转录组拷贝数分析结果解读和应用
生存分析
前面我们介绍了正向鉴定关键单细胞亚群的流程,就是首先降维聚类分群然后看哪个亚群是有临床意义,详见:
- 关键单细胞亚群辅助判定之生存分析
- 最简单的统计学之取交集
同时也给出来了反向鉴定关键单细胞亚群的流程,比如发表在2021年Nature Biotechnology上的Scissor算法,它们的结果非常一致,说明了算法的可靠性,而且类似的算法还有一个发表在NAR的一篇算法文章《scAB detects multiresolution cell states with clinical significance by integrating single-cell genomics and bulk sequencing data》,DOI10.1093/nar/gkac1109
其它软件算法的演示,代码会持续更新在这个链接哈,大家一定要抽空测试,链接: https://pan.baidu.com/s/1geW1MTLRizcJWEESdjMN7g?pwd=96t6