众所周知,2型糖尿病是一种复杂的代谢紊乱,会引起各种并发症,包括牙周炎。所以研究者们就选取了这两个疾病( Type 2 diabetes mellitus (DM)和 periodontitis (PD) )的患者的PBMC公共数据集做研究:
- GSE165816 includes the transcriptional profiles of PBMCs obtained from patients with DM and healthy donors.
- GSE164241 is a scRNA‐seq dataset of gingival tissues from patients with PD and from healthy controls.
两个单细胞转录组数据集汇总后是:
- 11 healthy controls,
- 10 patients with PD without DM,
- six patients with PDDM
值得一提的是这个数据挖掘是韩国人做的哦,标题是:《Immunological link between periodontitis and type 2 diabetes deciphered by single‐cell RNA analysis》
首先是降维聚类分群和分组后看比例变化
如下所示:
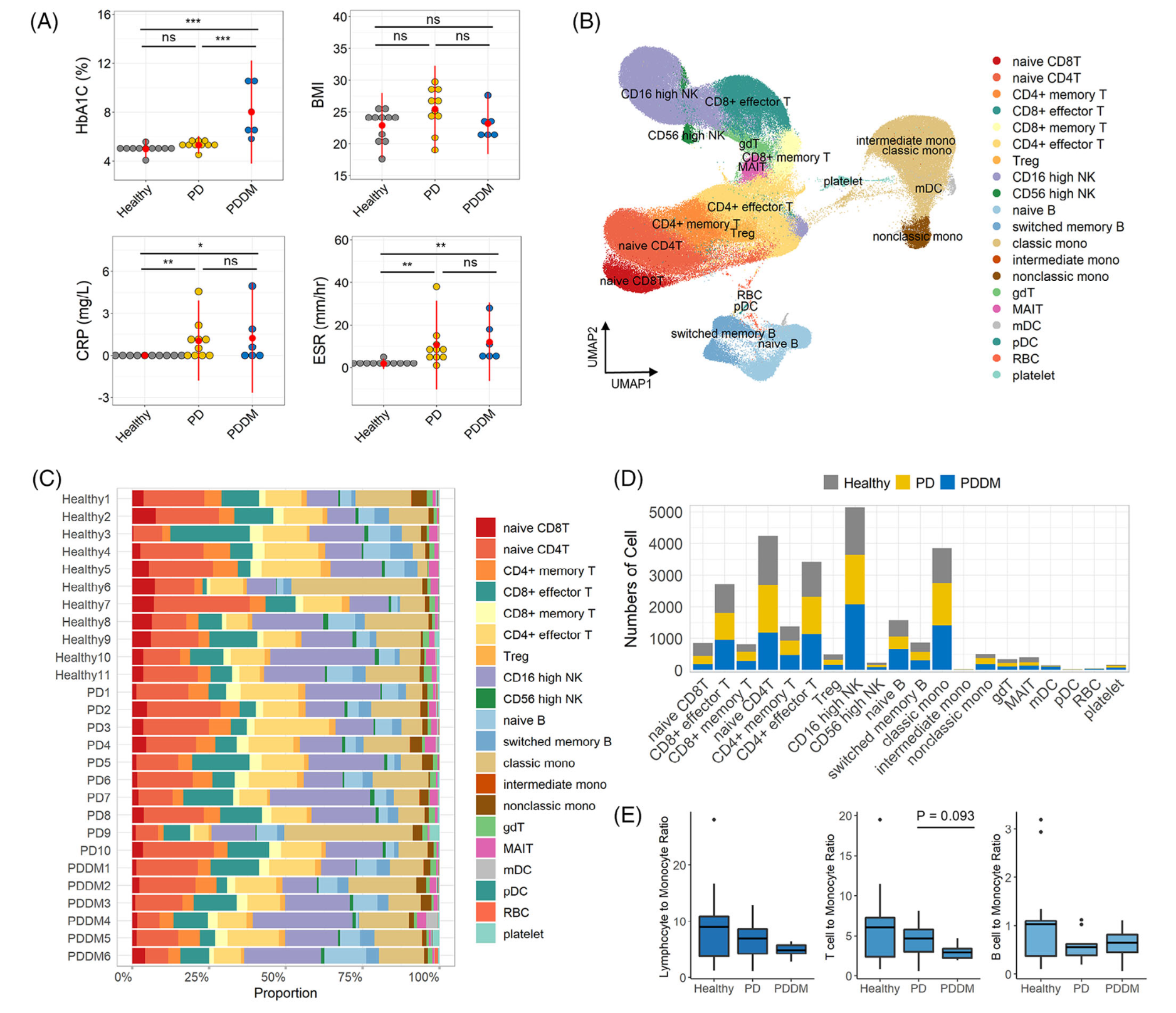
然后是各个单细胞亚群在不同分组的各种差异分析和富集分析
如下所示:
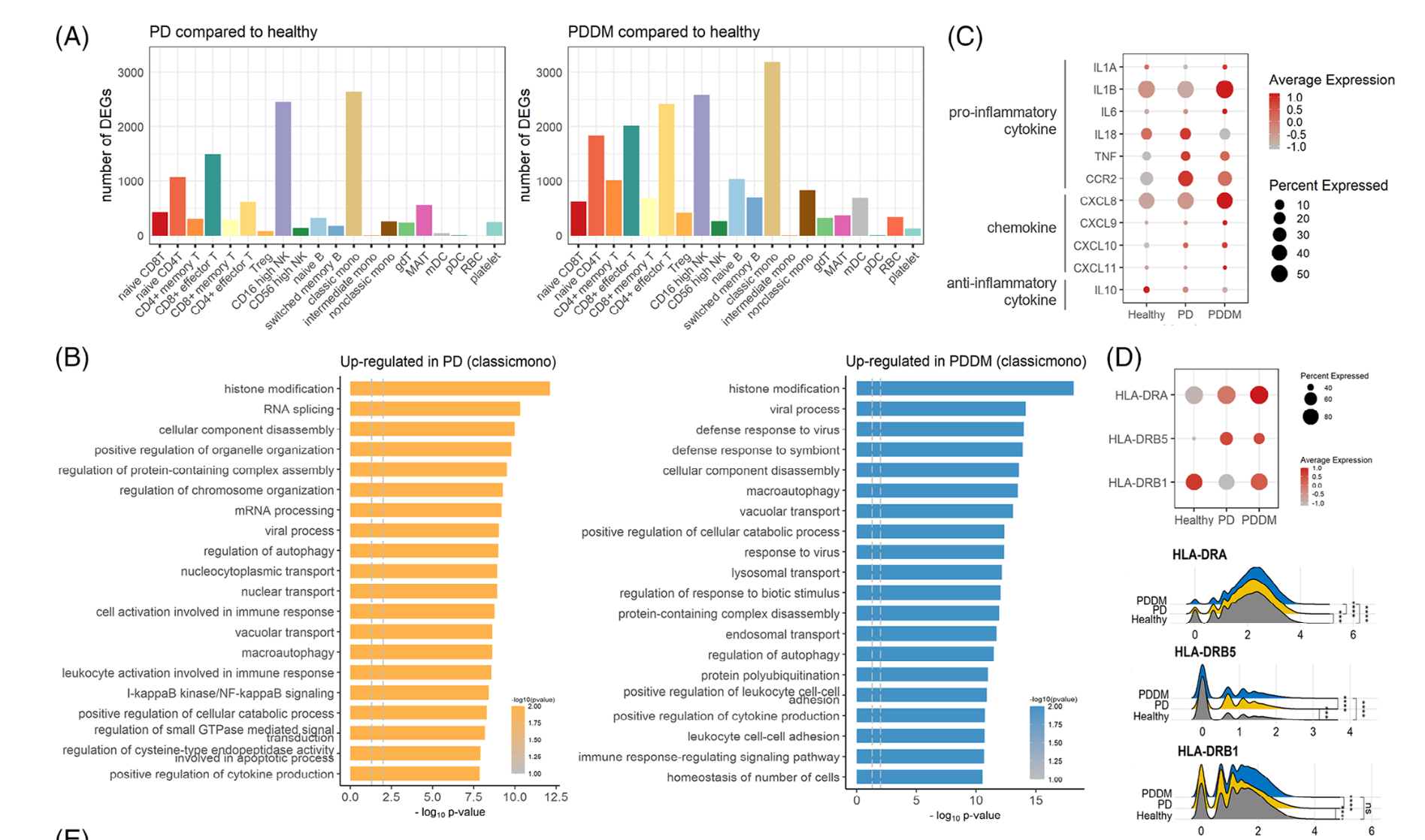
高级分析
主要是针对具体的某个细胞亚群看功能变化,比如这个文章就是针对 CD8T and NK cells
- 打分:The cytotoxicity, exhaustion and activity scores
- 拟时序
- 细胞通讯
首先看看GSE164241
是2021发表在CELL杂志的《Human oral mucosa cell atlas reveals a stromal- neutrophil axis regulating tissue immunity》,文章的第一层次降维聚类分群是比较简单的:
- endothelial (ACKR1, RAMP2, SELE, VWF, PECAM1),
- fibroblast (LUM, COL3A1, DCN, COL1A1, CFD),
- immune (CD69, CD52, CXCR4, PTPRC, HCST),
- epithelial (KRT14, KRT5, S100A2, CTSA, SPRR1B)
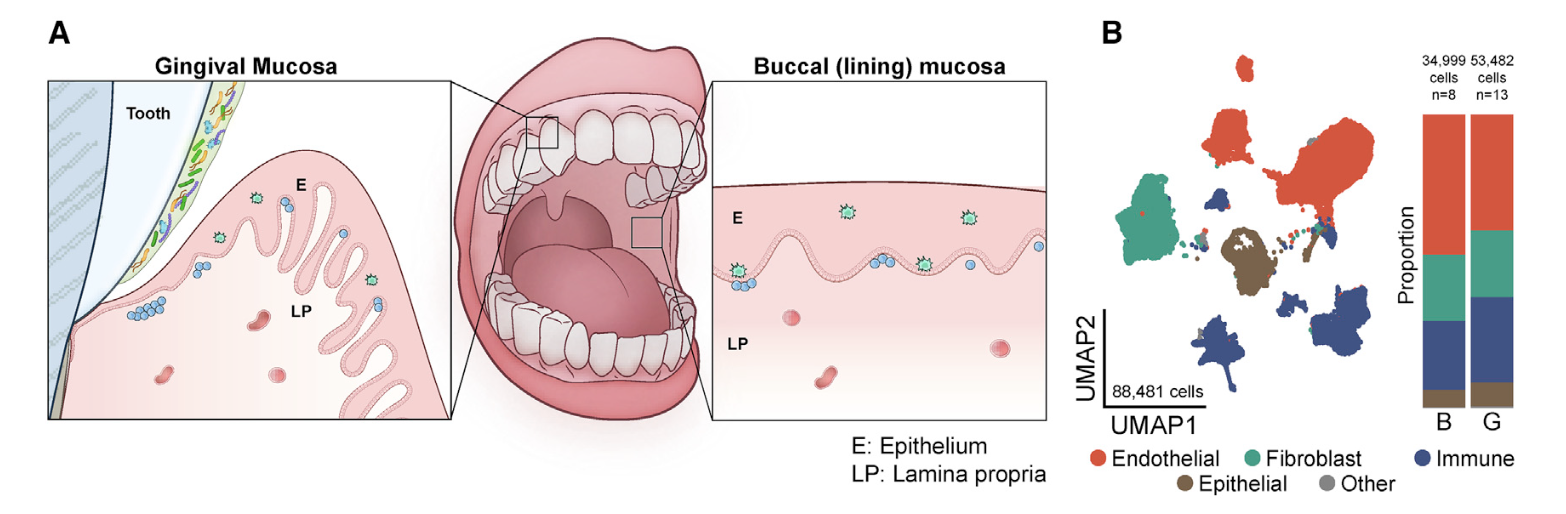
是标准的10x数据集,所以很容易导入到R里面进行分析
###### step1:导入数据 ######
samples=list.files('GSE164241_RAW/outputs/')
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
folder=file.path('GSE164241_RAW/outputs/',pro)
print(pro)
print(folder)
print(list.files(folder))
sce=CreateSeuratObject(counts = Read10X(folder),
project = pro ,
min.cells = 5,
min.features = 300)
return(sce)
})
names(sceList)
samples
names(sceList) = samples
sce.all <- merge(sceList[[1]], y= sceList[ -1 ] ,add.cell.ids = samples)
as.data.frame(sce.all@assays$RNA@counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all@meta.data$orig.ident)
library(stringr)
sce.all@meta.data$group = substring(sce.all@meta.data$orig.ident,1,2)
table(sce.all@meta.data$group )
head(sce.all@meta.data)
然后看GSE165816
是文章: Single cell transcriptomic landscape of diabetic foot ulcers. Nat Commun 2022 ,它的第一层次降维聚类分群要复杂一点,一步到位了:
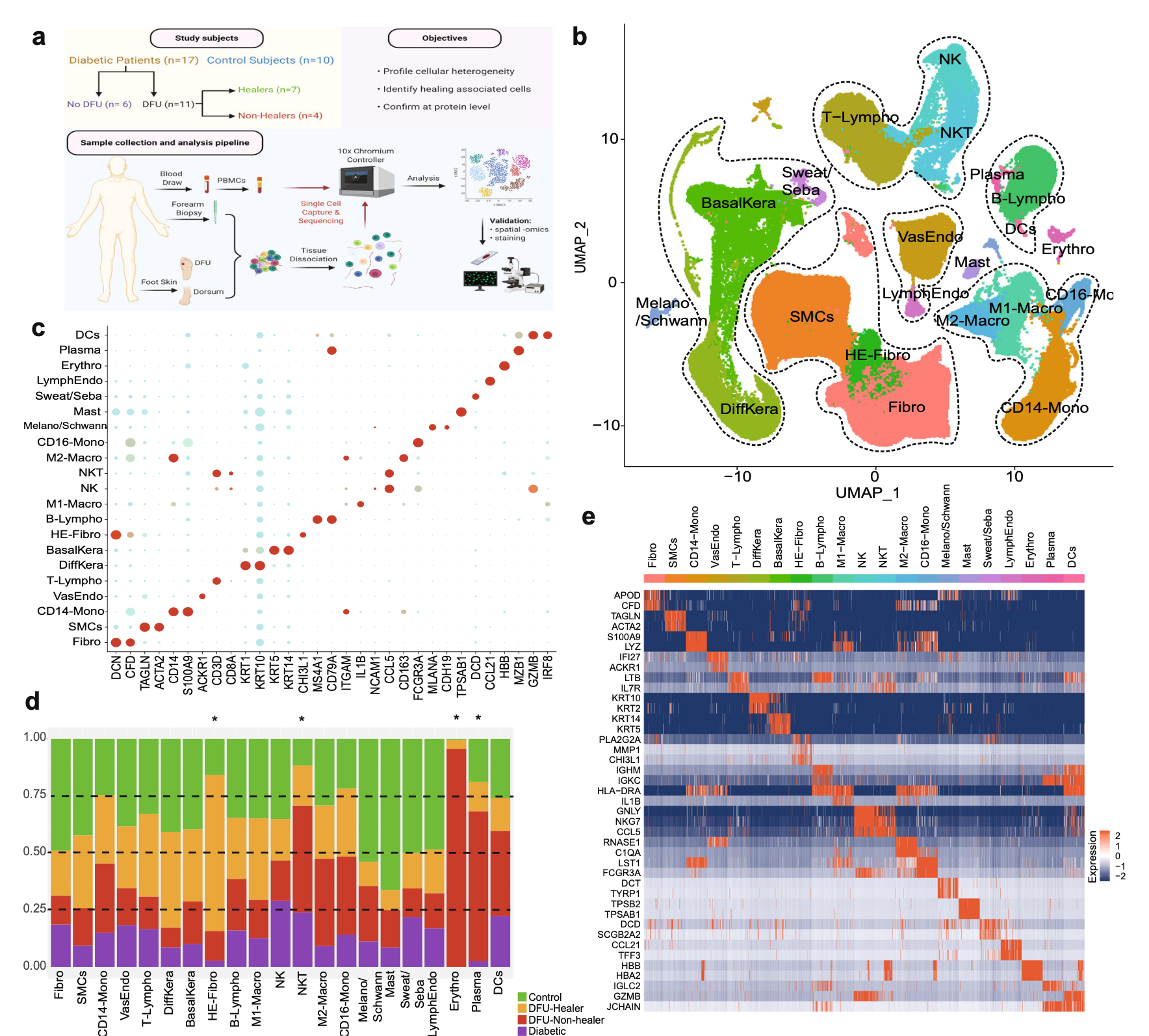
本质上也是读取文件即可,代码是:
library(data.table)
dir='GSE165816_RAW/'
samples=list.files( dir )
samples
library(data.table)
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
ct=fread(file.path( dir ,pro),data.table = F)
ct[1:4,1:4]
rownames(ct)=ct[,1]
ct=ct[,-1]
sce=CreateSeuratObject(counts = ct ,
project = gsub('counts.csv.gz','',
strsplit(pro,'_')[[1]][2]),
min.cells = 5,
min.features = 300,)
return(sce)
})
names(sceList)
samples = gsub('counts.csv.gz','',samples)
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples)
as.data.frame(sce.all@assays$RNA@counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all$orig.ident)
我的问题是?
这两个疾病的患者的PBMC跟正常人的在单细胞转录组水平差异很大吗?有必要这样做吗?
现在呢,基本上每个疾病都是有公开的单细胞数据集,而且很多疾病都是多个数据集,是不是可以做各种各样的联合分析了呢?