最近看到了《单细胞天地》公众号介绍了一个文章肿瘤病人的单细胞转录组数据文章,器标题:《Subclone-specific microenvironmental impact and drug response in refractory multiple myeloma revealed by single‐cell transcriptomics》
-
发表日期和杂志:2021年发表在Nature Communications上
其中第一层次降维聚类分群是:clustering and cell type identification was based on scRNA-seq of 212,404 cells from primary mononuclear bone marrow samples of RRMM patients (n = 20) as shown by an UMAP embedding colored by sample
without batch-effect correction.
然后呢,效果如下所示,感觉就有点奇怪,因为恰好是肿瘤细胞和正常细胞在umap图上面就泾渭分明成为了两个部分(下图的上半部分是肿瘤细胞,然后下半部分就是正常细胞):
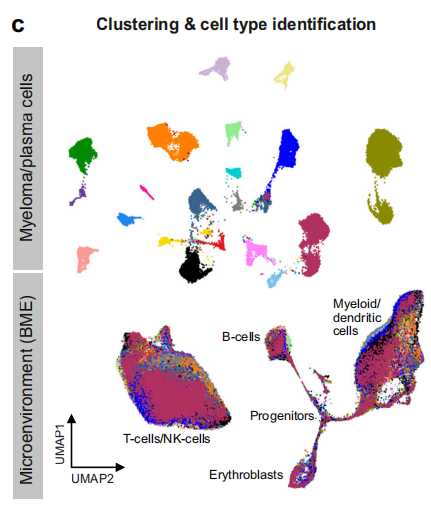
起初我是不太相信的,然后我就下载了这个数据集的GSE161801_RAW.tar压缩包然后解压后如下所示的文本文件:
$ ls -lh *gz|cut -d" " -f 5-
248K Nov 18 2020 GSM4914649_K43R_2FTMUU_T1.csv.gz
3.3M Nov 18 2020 GSM4914650_K43R_2FTMUU_N1.csv.gz
4.7M Nov 18 2020 GSM4914651_K43R_2FTMUU_T2.csv.gz
5.3M Nov 18 2020 GSM4914652_K43R_2FTMUU_N2.csv.gz
2.6M Nov 18 2020 GSM4914653_K43R_32ARQL_T2.csv.gz
3.6M Nov 18 2020 GSM4914654_K43R_32ARQL_N2.csv.gz
7.4M Nov 18 2020 GSM4914655_K43R_37A66E_T1.csv.gz
9.2M Nov 18 2020 GSM4914656_K43R_37A66E_N1.csv.gz
3.6M Nov 18 2020 GSM4914657_K43R_37A66E_T2.csv.gz
1.6M Nov 18 2020 GSM4914658_K43R_37A66E_N2.csv.gz
1.1M Nov 18 2020 GSM4914659_K43R_6K8YTY_T2.csv.gz
。。。。。。。。。。。。。。。。。
所以就批量读取它们,代码如下所示:
library(data.table)
dir='../GSE161801_RAW/'
samples=list.files( dir ,pattern = 'gz')
samples
library(data.table)
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
ct=fread(file.path( dir ,pro),data.table = F)
ct[1:4,1:4]
rownames(ct)=ct[,1]
ct=ct[,-1]
sce=CreateSeuratObject(counts = ct ,
project = gsub('matrix.txt.gz','',
strsplit(pro,'_')[[1]][3]) ,
min.cells = 5,
min.features = 300 )
table(sce$orig.ident)
sce$orig.ident = gsub('matrix.txt.gz','',
strsplit(pro,'_')[[1]][3])
return(sce)
})
names(sceList)
library(stringr)
samples=gsub('matrix.txt.gz','',str_split(samples,'_',simplify = T)[,3])
samples
names(sceList) = samples
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples)
as.data.frame(sce.all@assays$RNA@counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all$orig.ident)
一般情况下,大家会对上面的全部的样品的Seurat对象进行harmony整合,示例代码如下所示:
sce=sce.all
sce <- NormalizeData(sce,
normalization.method = "LogNormalize",
scale.factor = 1e4)
sce <- FindVariableFeatures(sce)
sce <- ScaleData(sce)
sce <- RunPCA(sce, features = VariableFeatures(object = sce))
if(F){
library(harmony)
seuratObj <- RunHarmony(sce, "orig.ident")
names(seuratObj@reductions)
seuratObj <- RunUMAP(seuratObj, dims = 1:15,
reduction = "harmony")
DimPlot(seuratObj,reduction = "umap",label=T )
sce=seuratObj
sce <- FindNeighbors(sce, reduction = "harmony",
dims = 1:15)
}
sce <- RunUMAP(sce, dims = 1:15 )
sce <- FindNeighbors(sce, dims = 1:15)
sce.all=sce
FeaturePlot(sce.all,raster = F,features = 'TNFRSF17')
但是我为了还原文章的结果,选择了放弃harmony整合后看降维聚类分群,然后可视化多发性骨髓瘤领域的特殊基因TNFRSF17的表达量,如下所示:
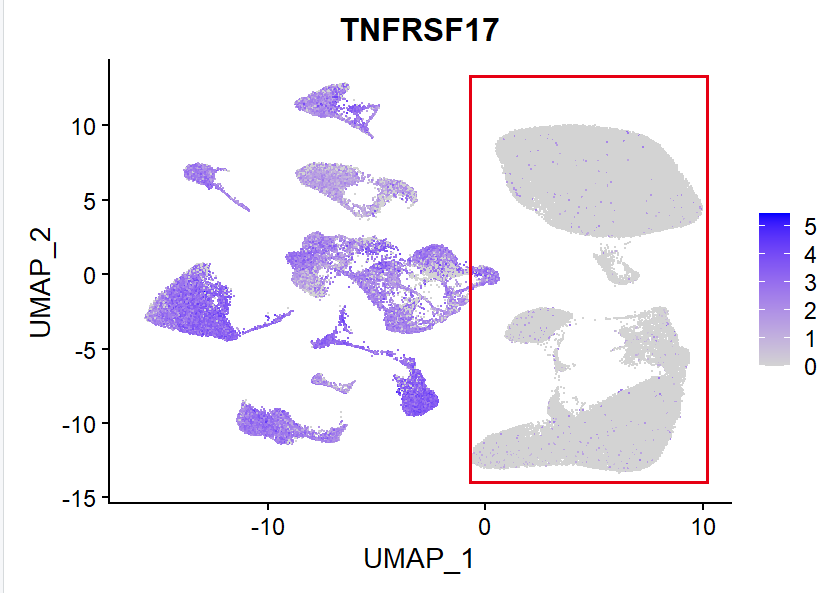
居然真的是恶性肿瘤细胞和正常的血液免疫细胞如此的泾渭分明,不是文章瞎搞,上面的UMAP图里面的左边部分全部是肿瘤细胞然后右边部分是正常细胞。而且通过常规基因我们可以给右边的正常的血液免疫细胞进行简单的命名,比如t细胞和b细胞还有髓系免疫细胞。
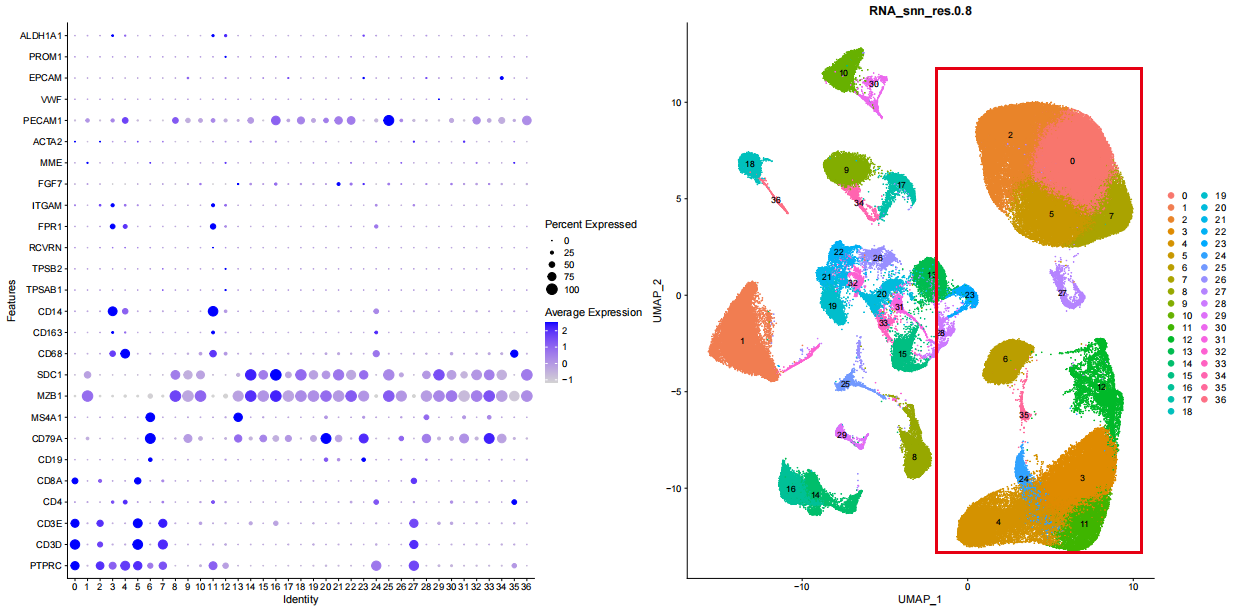
但是,如果我们这最开始的降维聚类分群流程里面的选择了对这些样品进行了harmony整合,就会出现很诡异的结果:
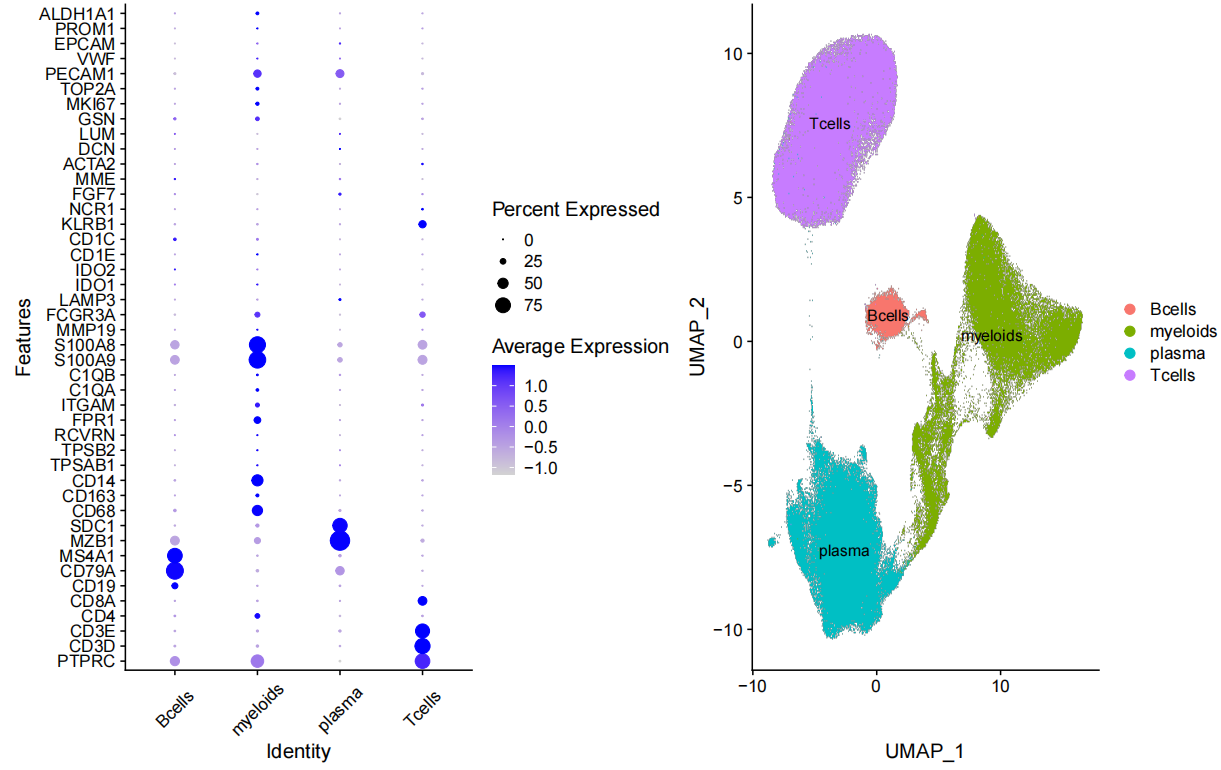
这个时候不要说是肿瘤病人异质性问题了,连肿瘤细胞和正常细胞都会被混合在一起,因为这个是血液肿瘤所以都是免疫细胞,它们这被harmony抹除样品个体差异的时候就顺便把肿瘤细胞和正常细胞的差异也给抹除了。
那么肿瘤病人应该是第一层次降维聚类分群就不harmony吗?
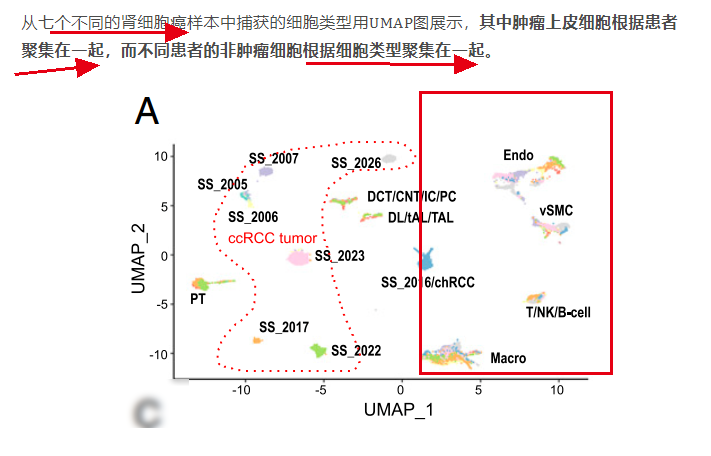
可以看到其它类似的肿瘤病人的单细胞转录组数据如果第一层次降维聚类分群就不harmony那么结果也是病人独立成群,比如《Single-cell analyses of renal cell cancers reveal insights into tumor microenvironment, cell of origin, and therapy response》也是如此
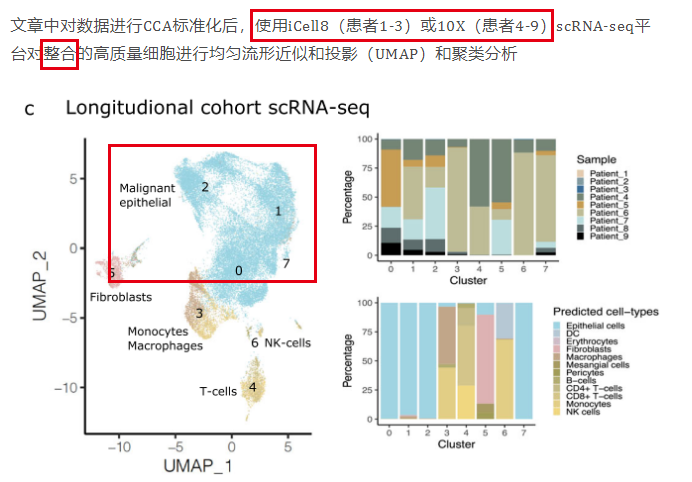
如果多个样品进行了整合,也就是说第一层次降维聚类分群采用了harmony,一般来说就不会有病人特异性独立成一个群的现象,比如《Evolution of core archetypal phenotypes in progressive high grade serous ovarian cancer》:
也就是说harmony在抹除病人异质性这方面的效果是杠杠的,比如2021的NC文章里面的25个病人也是被整合的非常干净:《An N-Cadherin 2 expressing epithelial cell subpopulation predicts response to surgery, chemotherapy and immunotherapy in bladder cancer》

上皮细胞里面的有一些细分亚群是恶性的肿瘤细胞,需要跑inferCNV流程才有可能区分哦,详见:肿瘤单细胞转录组拷贝数分析结果解读和应用,而且很明显这里面的降维聚类分群理论上是跟inferCNV流程的拷贝数冲突的,并不是说上面的5个亚群要么是恶性要么是正常二倍体,而是5个亚群本身在拷贝数变化层面仍然是有异质性!类似于下面的效果:

上面的画圈的两个亚群里面都是部分细胞是恶性的,而且有意思的是这两个降维聚类分群后的完全不同的亚群里面的部分细胞居然是拷贝数可以融合,这两个维度的分群是可以冲突的。
这个问题讨论很多次了其实
比如前些天在《生信技能树》的微信视频号做了一个肿瘤单细胞转录组的数据分析直播,文章是:《Delineating the dynamic evolution from preneoplasia to invasive lung adenocarcinoma by integrating single-cell RNA sequencing and spatial transcriptomics》详见:换一个分析策略会导致文章的全部论点都得推倒重来吗。
其中第一层次降维聚类分群后给出来了合理的生物学命名,详见:肿瘤单细胞转录组的第一层次降维聚类分群, 整体来说这个复现的代码在百度云分享给大家:链接:https://pan.baidu.com/s/1niFqyAiUU3yXK1W26b8RvQ?pwd=nbmj
这里面的第一层次降维聚类分群的时候多个病人样品就进行了harmony整合,值得注意是我们依赖于这个V4的版本的Seurat流程做出来了大量的公共数据集的单细胞转录组降维聚类分群流程,100多个公共单细胞数据集全部的处理,链接:https://pan.baidu.com/s/1MzfqW07P9ZqEA_URQ6rLbA?pwd=3heo
然后提取了上皮细胞后仍然是进行了harmony整合,仍然是可以有他们自己的生物学故事,因为上皮细胞harmony整合,可以很方便区分正常细胞和恶性细胞后,这个时候跑的是inferCNV流程,详见:肿瘤单细胞转录组拷贝数分析结果解读和应用
而且有正向鉴定关键单细胞亚群的流程,就是首先降维聚类分群然后看哪个亚群是有临床意义,详见:
同时也给出来了单细胞亚群的流程,比如发表在2021年Nature Biotechnology上的Scissor算法,它们的结果非常一致,说明了算法的可靠性,而且类似的算法还有一个发表在NAR的一篇算法文章《scAB detects multiresolution cell states with clinical significance by integrating single-cell genomics and bulk sequencing data》,DOI10.1093/nar/gkac1109,其它软件算法的演示,代码会持续更新在这个链接哈,大家一定要抽空测试,链接: https://pan.baidu.com/s/1geW1MTLRizcJWEESdjMN7g?pwd=96t6