如果大家跑过inferCNV这样的肿瘤单细胞拷贝数变异代码,就会发现它这个函数(CreateInfercnvObject),有一个诡异的设置,就是它会自动读取当前工作目录下面的3个文件 :
expFile='expFile.txt'
groupFiles='groupFiles.txt'
geneFile='geneFile.txt'
# duplicate 'row.names' are not allowed
library(infercnv)
infercnv_obj = CreateInfercnvObject(raw_counts_matrix=expFile,
annotations_file=groupFiles,
delim="\t",
gene_order_file= geneFile,
ref_group_names=c('ref-1',
'ref-2')) ## 这个取决于自己的分组信息里面的
这样的话, 我们就不得不把在R里面的变量,写出到硬盘,成为文件。感觉是在使用一个独立的网页工具。。。。
如下所示,是我以前的代码;
dim(dat)
expFile='expFile.txt'
write.table(dat,file = expFile,sep = '\t',quote = F)
groupFiles='groupFiles.txt'
head(groupinfo)
write.table(groupinfo,file = groupFiles,sep = '\t',quote = F,col.names = F,row.names = F)
head(geneInfor)
geneFile='geneFile.txt'
write.table(geneInfor,file = geneFile,sep = '\t',quote = F,col.names = F,row.names = F)
其中,dat变量是单细胞表达量矩阵,通常是几万个细胞,几万个基因,这样的话,输出的txt文本文件会超级大,非常的耗时!!!是时候改变它了,所以我求助了chatGPT,如果大家不能使用外网的chatGPT,也可以参考我们通过BioinfoArk提供的中国区chatGPT的查询方式:
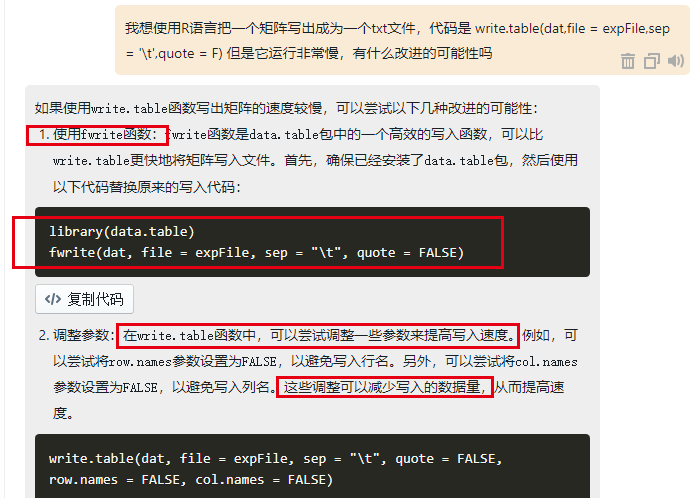
我采纳了它的第一个建议,然后试着对比一下:
# 代码段1
start_time1 <- Sys.time()
# Your code here
write.table(dat,file = expFile,sep = '\t',quote = F)
end_time1 <- Sys.time()
execution_time1 <- end_time1 - start_time1
# 代码段2
start_time2 <- Sys.time()
# Your code here
fwrite(dat, file = expFile, row.names = T,sep = "\t", quote = FALSE)
end_time2 <- Sys.time()
execution_time2 <- end_time2 - start_time2
# 比较耗时
if (execution_time1 < execution_time2) {
cat("代码段1执行时间更短:", execution_time1, "\n")
} else if (execution_time1 > execution_time2) {
cat("代码段2执行时间更短:", execution_time2, "\n")
} else {
cat("两个代码段执行时间相同:", execution_time1, "\n")
}
这个时间层面的提高,数值提高简直是恐怖如斯!!!!
代码段2执行时间更短: 0.392374
> execution_time1
Time difference of 1.92164 mins