早在2021的CELL文章:《Atlas of clinically distinct cell states and ecosystems across human solid tumors》就介绍了这个EcoTyper,它能根据肿瘤的单细胞转录组数据集的降维聚类分群结果,去以前常规的bulk转录组数据集(包括表达量芯片和转录组测序)里面去推断各个单细胞亚群比例后汇总成为不同的细胞状态组合情况以及多细胞群落情况。
那个2021的CELL文章的作者为了评估EcoTyper定义的71种细胞状态的保真度,他们探究了4种人类癌症的200,000个单细胞转录组中每种状态的存在情况。在scRNA-seq数据中,94%的细胞状态(71个中的67个)可显著恢复,而且无论平台、细胞类型或数据集如何,恢复率都很高,突显了结果的可靠性。并且绘制了15,008例肿瘤中69种细胞状态的预后图。在发现队列调查的16种上皮癌类型中,大多数细胞状态(69种中的39种)与总生存率显著相关。
既然这些细胞状态组合情况以及多细胞群落情况,在肿瘤里面是跟预后会有关系, 那么毫无疑问,每个癌症都是可以做同样的数据分析啦。比如2023年7月的文章:《Deep immunophenotyping reveals clinically distinct cellular states and ecosystems in large-scale colorectal cancer》就是收集了14个GEO数据集1725个CRC样本和TCGA的625个样本,样品数量很大,如下所示:

但凡是做过自己疾病的背景调研,很容易收集到几十个甚至上百个常规的bulk转录组数据集(包括表达量芯片和转录组测序),而且绝大部分数据集都被反反复复挖掘过一次啦。
同理,任意疾病的单细胞数据集那也是几十个以上了,比如这个文章就是取了 GSE132465 (n = 23) 和 E-MTAB-8107 (n = 7)这两个数据集 ,进行降维聚类分群:
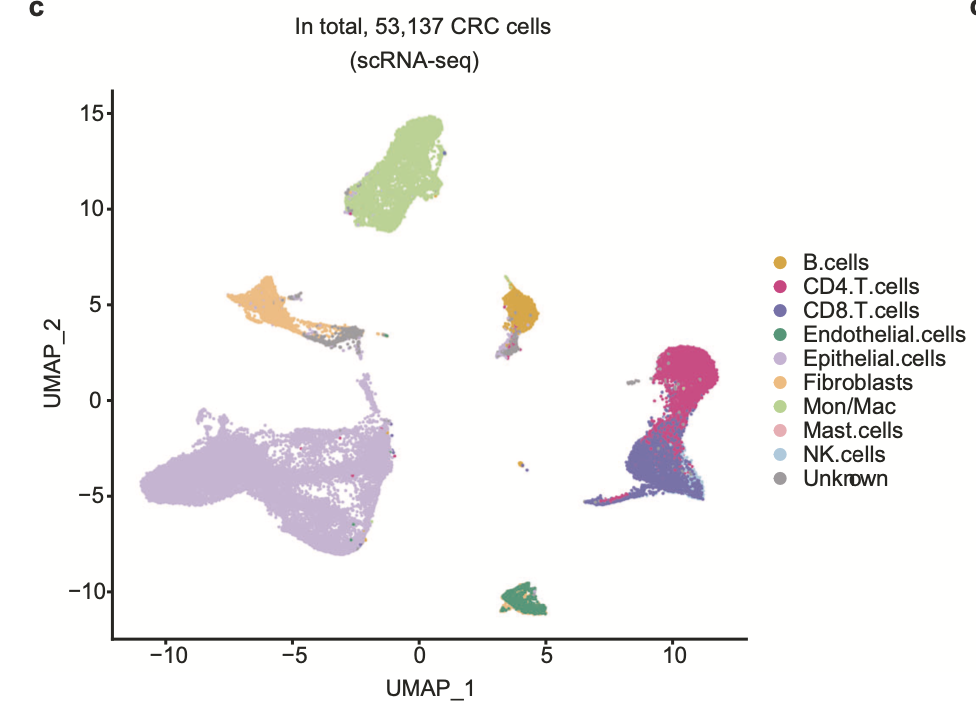
这样的单细胞转录组数据分析的标准降维聚类分群,并且进行生物学注释后的结果。可以参考前面的例子:人人都能学会的单细胞聚类分群注释 ,我们演示了第一层次的分群。如果你对单细胞数据分析还没有基础认知,可以看基础10讲:
- 01. 上游分析流程
- 02.课题多少个样品,测序数据量如何
- 03. 过滤不合格细胞和基因(数据质控很重要)
- 04. 过滤线粒体核糖体基因
- 05. 去除细胞效应和基因效应
- 06.单细胞转录组数据的降维聚类分群
- 07.单细胞转录组数据处理之细胞亚群注释
- 08.把拿到的亚群进行更细致的分群
- 09.单细胞转录组数据处理之细胞亚群比例比较
这个文章非常优秀提供了全部的代码
- The codes of EcoTyper were obtained from https://ecotyper.stanford.edu/carcinoma/.
- Other codes used in this study are available from https://github.com/ComputationalEpigeneticsLab/Tumor-cellular-states-and-ecosystems-in-CRC
比如我们打开作者的代码,如下所示:
60 Distribution of cellular states in CEs.R
211 Hallmark+immune enrichment analysis.R
198 Molecular characteristics of seven CEs.R
78 TFs predict clinical survival.R
354 TFs regulating the genes.R
83 The GO enrichment analysis.R
43 The analysis of 198 drugs.R
108 The expression of TF.R
71 The expression of fib-gene.R
460 The plot of GSEA.R
37 The proportion of CRC patients recovered in validation cohorts.R
35 The proportions of cell types in CEs.R
28 The proportions of patients.R
24 UMAP projection of cellular states.R
可以看到绝大部分代码都不复杂,主要就是可视化,部分代码相信大家肯定是用得到,比如:
#### sankey
library(ggalluvial)
library(RColorBrewer)
library(randomcoloR)
col_sankey<-c('B.cells'="#E6AB02", 'CD4.T.cells'="#E7298A", 'CD8.T.cells'="#7570B3",
'Dendritic.cells'="#D95F02",'Endothelial.cells'="#1B9E77",'Epithelial.cells'="#CAB2D6",
'Fibroblasts'="#FDBF6F",'Mast.cells'="#F7AEB3",
'Monocytes.and.Macrophages'="#B2DF8A",'NK.cells'="#A6CEE3",'PCs'="#999999",'PMNs'="#F780BF",
'E1'="#D6372E",'E2'="#FADD4B",'E3'="#70B460",'E4'="#E690C1",
'E5'="#985EA8",'E6'="#A3A3A3",'E7'="#B7D3E5")
pdf("state_ecotype_sankey2.pdf",width = 6,height = 8)
ggplot(state_ecotype, aes(x = variable, y = link,
stratum = value, alluvium = flow, fill = value)) +
geom_stratum() +
geom_flow(aes.flow = 'forward') +
scale_fill_manual(values = col_sankey[rank(1:44)]) +
guides(fill=FALSE)+
geom_text(stat = 'stratum', infer.label = TRUE, size = 2.5) +
labs(x = '', y = '') +
theme(legend.position = 'none', panel.background = element_blank(),
line = element_blank(), axis.text.y = element_blank())+
scale_x_discrete(limits = c('CellType', 'Ecotype'))
dev.off()
得到如下所示的右图(sankey):
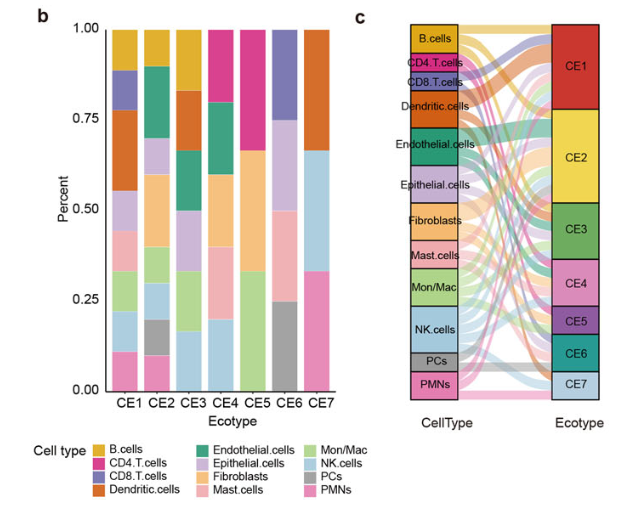
还等什么呢
在任意癌症上面复现吧,数据处理的过程中,相信大家就有会有自己的不一样的生物学故事。比如上面的文章侧重于肿瘤成纤维,大家可以侧重于其它单细胞亚群,
通常我们拿到了肿瘤相关的单细胞转录组的表达量矩阵后的第一层次降维聚类分群通常是:
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibro or CD31+,PECAM1,endo)
参考我前面介绍过 CNS图表复现08—肿瘤单细胞数据第一次分群通用规则,这3大单细胞亚群构成了肿瘤免疫微环境的复杂。绝大部分文章都是抓住免疫细胞亚群进行细分,包括淋巴系(T,B,NK细胞)和髓系(单核,树突,巨噬,粒细胞)的两大类作为第二次细分亚群。但是也有不少文章是抓住stromal 里面的 fibro 和endo进行细分,并且编造生物学故事的。
前面我们已经介绍了心肝脾肺肾等多个器官的上皮细胞的细分亚群, 以及免疫细胞里面的髓系和B细胞细分亚群:
其中髓系免疫细胞细分亚群也可以看张泽民课题组的泛癌层面数据挖掘文章,2021年2月发表在CELL的《A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells》
值得注意的是这个EcoTyper工具不好用哦
作者还构建的是shiny网页工具,而不是公开的R包方便大家使用,详细的链接是: https://ecotyper.stanford.edu/carcinoma/#shiny-tab-explore
有体验 过这个工具的小伙伴可以分享一下使用心得哈, 我感觉这个思路其实很简单的, 就是任意去卷积软件把单细胞转录组降维聚类分群的信息拿去常规的bulk转录组数据集(包括表达量芯片和转录组测序)去推断各个单细胞亚群比例后,使用这个比例信息进行分子分型即可!