官网说的很清楚:https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/fastq-input
To serve as inputs for cellranger, FASTQ files should conform to the naming conventions of bcl2fastq and mkfastq:
[Sample Name]S1_L00[Lane Number][Read Type]_001.fastq.gz
Where Read Type is one of:
I1: Sample index read (optional)I2: Sample index read (optional)R1: Read 1R2: Read 2
也就是说,其实跑他们自己的 cellranger 流程,我们只需要准备r1和r2文件即可。即使是这样,也有很多人会准备错误,正常准备好了FASTQ文件后走cellranger的定量流程即可,代码我已经是多次分享了。参考:- 10X单细胞转录组原始测序数据的Cell Ranger流程(仅需800元)
- 10X的单细胞转录组原始数据也可以在EBI下载
- 一个10x单细胞转录组项目从fastq到细胞亚群
- 一文打通单细胞上游:从软件部署到上游分析
- PRJNA713302这个10x单细胞fastq实战
- 一次曲折且昂贵的单细胞公共数据获取与上游处理
- 只能下载bam文件的10x单细胞转录组项目数据处理
- 不知道10x单细胞转录组样品和fastq文件的对应关系
- 10X单细胞转录组测序数据的 SRA转fastq踩坑那些事
- 10x的单细胞转录组fastq文件的R1和R2不能弄混哦
差不多几个小时就可以完成全部的样品的cellranger的定量流程。但是很多时候,大家是 从公共数据库下载的10x技术单细胞转录组测序数据,而不是自己的测序仪产出的数据,就容易出现,10x的单细胞转录组fastq文件的R1和R2不能弄混哦
比如:https://kb.10xgenomics.com/hc/en-us/articles/115003802691-How-do-I-prepare-Sequence-Read-Archive-SRA-data-from-NCBI-for-Cell-Ranger- ,需要注意的是使用参数--split-files来替代--split-3,就可以生成三个文件。
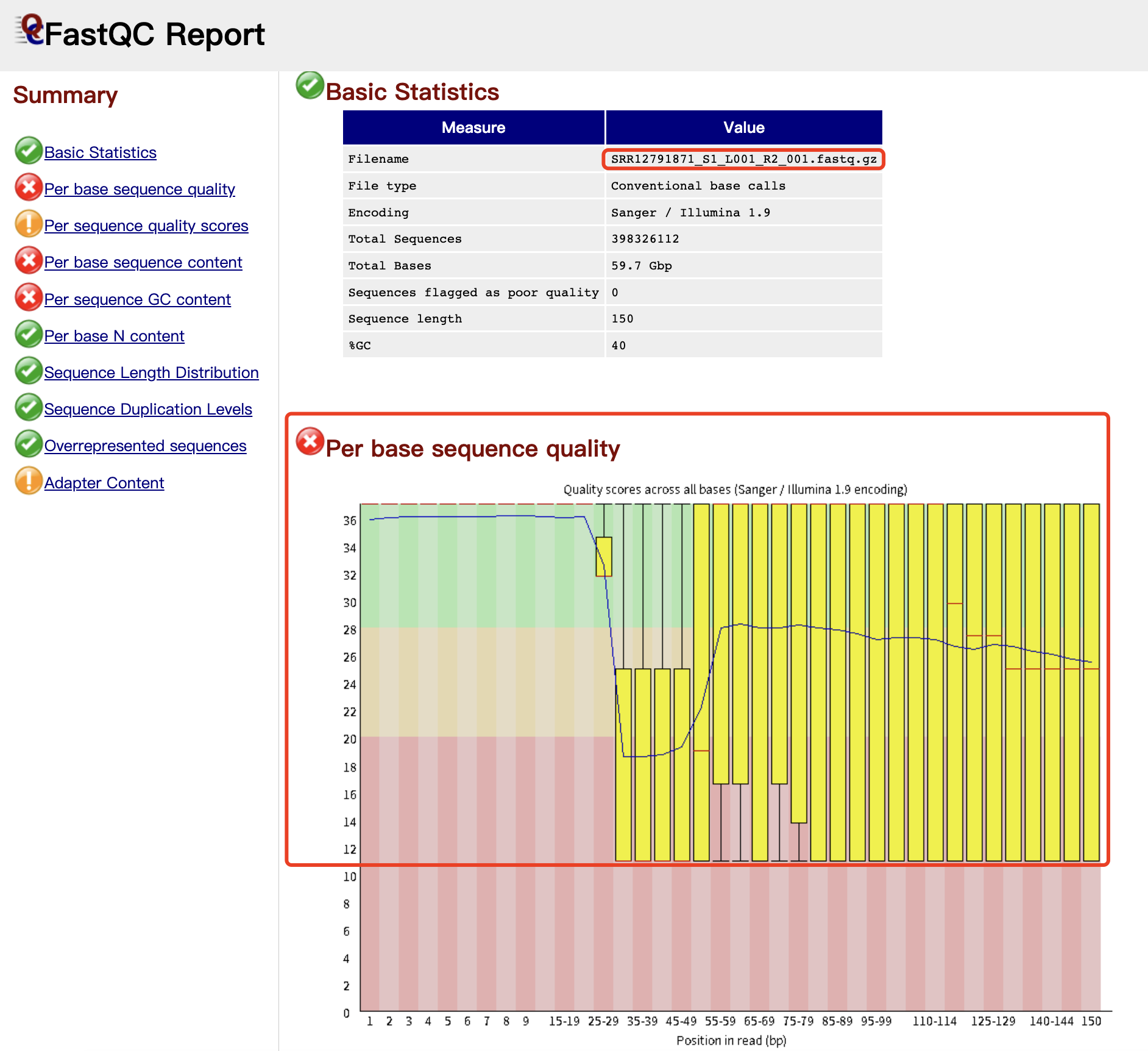
我们推荐的是对自己的准备好了的FASTQ文件跑一下fastqc软件,如下所示的就是命名错误啦 :
首先,1-26个cycle就是测序得到了26个碱基,先是16个Barcode碱基,然后是10个UMI碱基;这个文件就是R1了,但是它有可能也是在100bp或者150bp里面,因为测序仪就是这样的规格,只能说浪费掉。。。。
然后,27-34这8个cycle得到了8个碱基,就是i7的sample index;这个文件可有可无,就不关心它了。
最后35-132个cycle得到了98个碱基,就是转录本reads,也有可能是150bp长度的碱基啦,取决于测序仪规格,这个时候测序仪就充分利用了,不浪费。
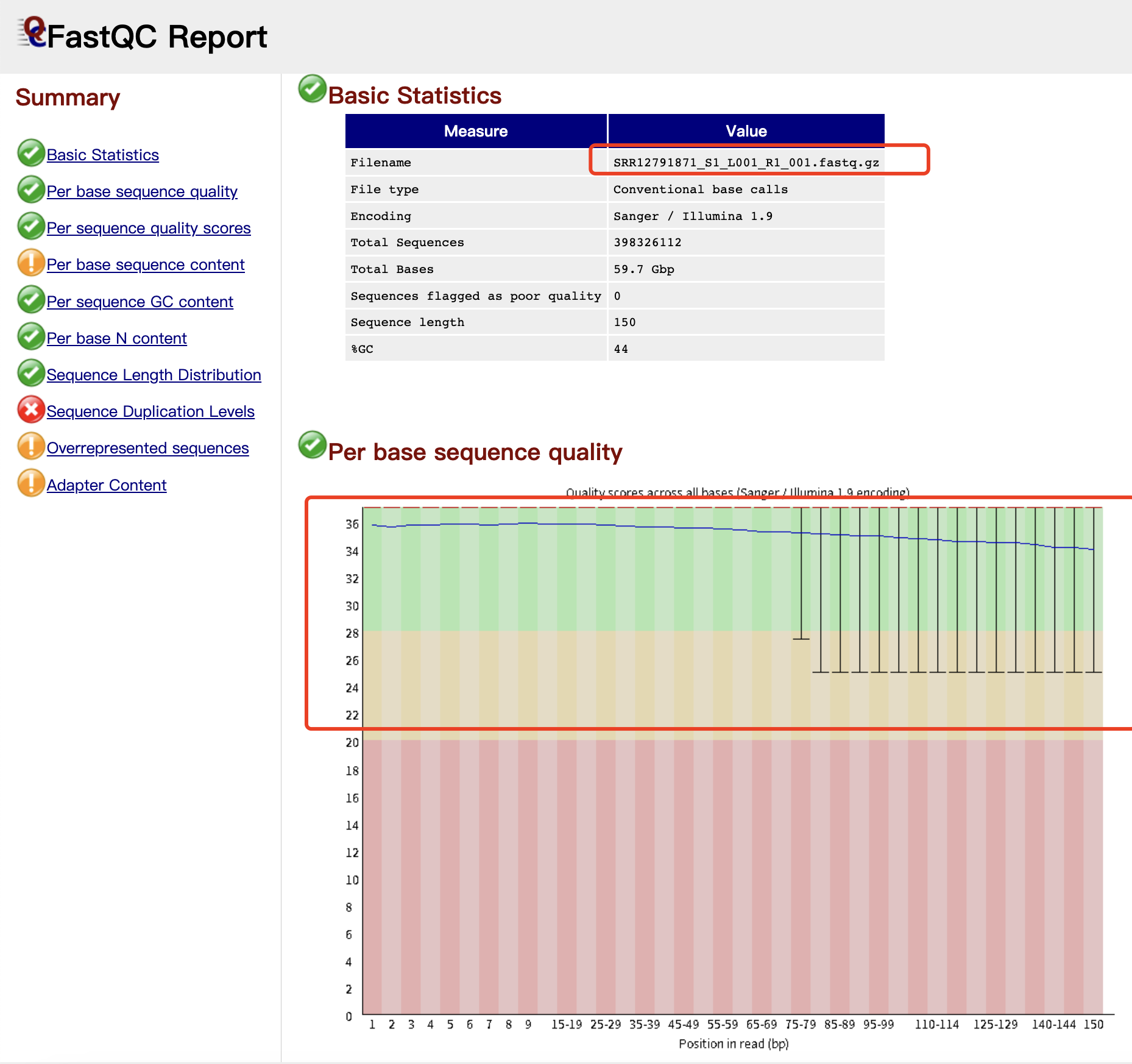
也就是说大家看得到r1文件其实有可能是很小也有可能是跟r2差不多大小,这样的话就会给大家带来困扰,简单的肉眼看两个准备好了的FASTQ文件的碱基长度是不够的,还需要看质量。如下所示的r2文件被弄错了成为了r1,所以r1文件也会被弄错了成为了r2,如下所示:
但是,我明明是给小伙伴们解释清楚了,但是仍然是有“好奇宝宝”不满意这个测序仪的浪费,认为明明是r1里面的碱基数量那么少,为什么要在150bp里面呢,希望自己切除它:
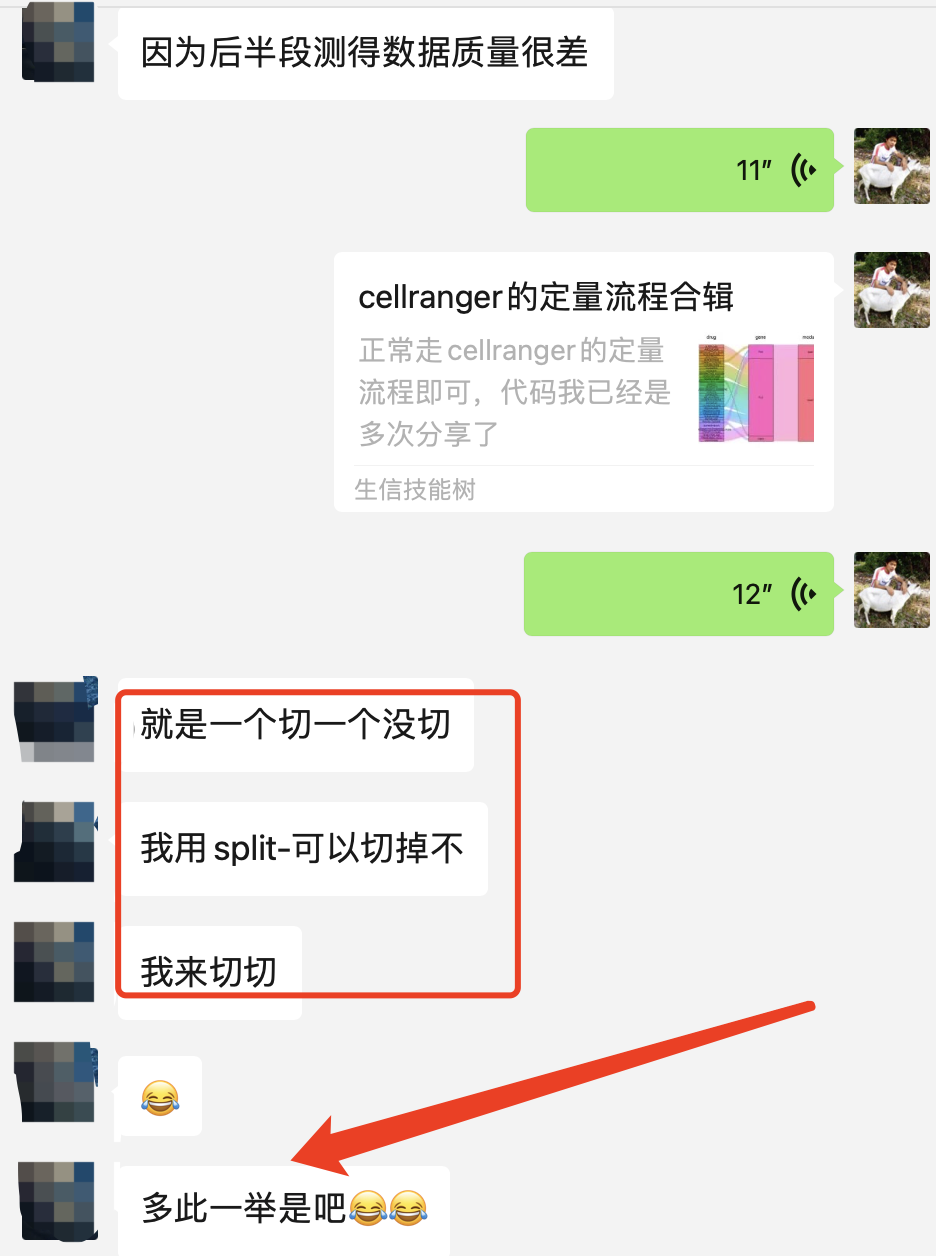
其实cellranger软件本身是有这个功能的, 完全没有必要自己提前处理r1的fq文件,不过呢,这个也确实是值得探索。参考:https://kb.10xgenomics.com/hc/en-us/articles/13179523030925-Why-do-I-have-an-alert-in-my-web-summary-with-Low-Fraction-of-Valid-UMIs-学徒作业
如果是需要你来修剪这个r1的fq文件,如何精准的保证它从测序仪出来的150bp变成16个Barcode碱基加上10个或者12个UMI碱基的短小的fq文件呢?