前面我们已经分享过一次10x技术空间单细胞上游定量案例,详见: 空间单细胞的上游定量流程(spaceranger,10x技术), 不过这样的分析理论上需要跑四五次才能算是完全掌握。所以我们接下来分享E-MTAB-12043数据集的:
E-MTAB-12043 是2023年初的数据:《RNA Spatial Sequencing of Colorectal Liver Metastases regarding their Histopathological Growth Patterns》,在线链接可以看:
- https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-12043
- https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-12043/sdrf?full=true
可以看到是6个样品的10x技术空间单细胞,但是有24次双端测序,所以是48个数据文件。而且是很标准的10x技术的空间单细胞转录组:

其中6个图片的路径是:
https://www.ebi.ac.uk/biostudies/files/E-MTAB-12043/Sample1.tif
https://www.ebi.ac.uk/biostudies/files/E-MTAB-12043/Sample3.tif
https://www.ebi.ac.uk/biostudies/files/E-MTAB-12043/Sample4.tif
https://www.ebi.ac.uk/biostudies/files/E-MTAB-12043/Sample5.tif
https://www.ebi.ac.uk/biostudies/files/E-MTAB-12043/Sample6.tif
https://www.ebi.ac.uk/biostudies/files/E-MTAB-12043/Sample7.tif
根据上面的链接很容易下载后可以看到:
ls -lh *tif|cut -d" " -f 5-
213M 8月 9 2022 Sample1.tif
211M 8月 9 2022 Sample3.tif
214M 8月 9 2022 Sample4.tif
198M 8月 9 2022 Sample5.tif
198M 8月 9 2022 Sample6.tif
195M 8月 9 2022 Sample7.tif
虽然说前些天我们演示了如何下载这个项目的fastq格式的测序数据原始文件,详见:aspera的高速下载确实很快吗,但是这个项目似乎是并没有给出来高速下载链接,所以只能说是传统下载方式了。然后是看看每个样品的fq文件,如下所示:
ls -lh *gz|cut -d" " -f 5-
1.1G 3月 4 01:14 Sample1_L1_1.fq.gz
1.6G 3月 4 01:21 Sample1_L1_2.fq.gz
1.2G 3月 4 04:02 Sample1_L2_1.fq.gz
1.7G 3月 4 04:07 Sample1_L2_2.fq.gz
1.2G 3月 4 02:48 Sample1_L3_1.fq.gz
1.8G 3月 4 02:53 Sample1_L3_2.fq.gz
1.1G 3月 4 02:56 Sample1_L4_1.fq.gz
1.7G 3月 4 03:01 Sample1_L4_2.fq.gz
1.5G 3月 3 22:11 Sample3_L1_1.fq.gz
2.2G 3月 3 23:03 Sample3_L1_2.fq.gz
1.6G 3月 4 01:48 Sample3_L2_1.fq.gz
2.3G 3月 4 01:55 Sample3_L2_2.fq.gz
1.6G 3月 4 03:25 Sample3_L3_1.fq.gz
2.3G 3月 4 03:31 Sample3_L3_2.fq.gz
1.5G 3月 4 02:00 Sample3_L4_1.fq.gz
2.3G 3月 4 02:07 Sample3_L4_2.fq.gz
有了这些文件,就可以开始我们的10x技术空间单细胞上游定量流程啦,从fastq文件开始, 走spaceranger定量流程,详情请见:空间单细胞的上游定量流程(spaceranger,10x技术)。我们来实战演练,首先是每个样品只需要是有r1和r2即可,并不一定要四个文件或者三个文件哈。但是文件名字一定要符合规则哦!
for file in *_1.fq.gz; do
new_name=$(echo "$file" | sed 's/_L/_S/'| sed 's/_1.fq.gz/_L001_R1_001.fastq.gz/')
ln "$file" "$new_name"
done
for file in *_2.fq.gz; do
new_name=$(echo "$file" | sed 's/_L/_S/'| sed 's/_2.fq.gz/_L001_R2_001.fastq.gz/')
ln "$file" "$new_name"
done
可以看到改名后的如下所示:
ls -lh *gz|cut -d" " -f 5-
1.1G 3月 4 01:14 Sample1_S1_L001_R1_001.fastq.gz
1.6G 3月 4 01:21 Sample1_S1_L001_R2_001.fastq.gz
1.2G 3月 4 04:02 Sample1_S2_L001_R1_001.fastq.gz
1.7G 3月 4 04:07 Sample1_S2_L001_R2_001.fastq.gz
1.2G 3月 4 02:48 Sample1_S3_L001_R1_001.fastq.gz
1.8G 3月 4 02:53 Sample1_S3_L001_R2_001.fastq.gz
1.1G 3月 4 02:56 Sample1_S4_L001_R1_001.fastq.gz
1.7G 3月 4 03:01 Sample1_S4_L001_R2_001.fastq.gz
1.5G 3月 3 22:11 Sample3_S1_L001_R1_001.fastq.gz
2.2G 3月 3 23:03 Sample3_S1_L001_R2_001.fastq.gz
1.6G 3月 4 01:48 Sample3_S2_L001_R1_001.fastq.gz
2.3G 3月 4 01:55 Sample3_S2_L001_R2_001.fastq.gz
1.6G 3月 4 03:25 Sample3_S3_L001_R1_001.fastq.gz
2.3G 3月 4 03:31 Sample3_S3_L001_R2_001.fastq.gz
1.5G 3月 4 02:00 Sample3_S4_L001_R1_001.fastq.gz
2.3G 3月 4 02:07 Sample3_S4_L001_R2_001.fastq.gz
写一个脚本文本文件( run_sapceranger.sh ),如下所示:
#! /bin/bash -xe
##
bin=/home/jmzeng/x10/pipeline/spaceranger-2.1.1/spaceranger
db=/home/jmzeng/x10/pipeline/refdata-gex-GRCh38-2020-A
ls $bin $db
fq_dir=${2}
/usr/bin/time -v $bin count --id=${1} \
--transcriptome=$db \
--fastqs=$fq_dir \
--sample=${1} \
--image=${3} \
--unknown-slide visium-1 \
--localcores=2 \
--localmem=80
上面的脚本文本文件( run_sapceranger.sh ),可以接受3个参数,包括样品的fastq格式的测序数据文件前缀,样品的fastq格式的测序数据文件所在的文件夹路径,以及样品的图片。我们来运行第一个样品,就是单个样品跑上面的脚本文本文件( run_sapceranger.sh )的代码:
mkdir rawdata
mv *.fastq.gz rawdata/
nohup bash run_sapceranger.sh Sample1 \
./rawdata \
./Sample1.tif \
1>log_Sample1.txt 2>&1 &
我们是多个样品有规则的文件,所以可以写一个批处理:
for i in {1..7}; do
nohup bash run_sapceranger.sh "Sample${i}" ./rawdata "./Sample${i}.tif" 1> "log_Sample${i}.txt" 2>&1 &
done
当然了,这一切代码成功运行的前提是,文件是齐全并且完整的,空间单细胞转录组需要图片和fastq文件哦! 这些文件都很大, 所以很容易下载失败,详见:aspera的高速下载确实很快吗。我这里也是遇到了4个文件下载失败的问题,我们针对全部的文件进行 gzip -t 的检验,发现是如下所示的4个文件失败了:
gzip: Sample1_S1_L001_R1_001.fastq.gz: invalid compressed data--crc error
gzip: Sample7_S2_L001_R2_001.fastq.gz: invalid compressed data--crc error
gzip: Sample7_S4_L001_R1_001.fastq.gz: invalid compressed data--crc error
gzip: Sample7_S4_L001_R2_001.fastq.gz: invalid compressed data--crc error
可以查看这失败的4个文件的md5,结果如下所示:
5a0fa0fbed1240bdd976f971ddb0d104 ../rawdata/Sample1_S1_L001_R1_001.fastq.gz
dafec34a3c656dca347d7111f45d9488 ../rawdata/Sample7_S2_L001_R2_001.fastq.gz
6c88e1c352f1d7816a8c5001f70daeef ../rawdata/Sample7_S4_L001_R1_001.fastq.gz
d1748a4944b6ada9c5fc5ab56f1eebd5 ../rawdata/Sample7_S4_L001_R2_001.fastq.gz
然后就重新下载了失败的这4个,并且再次查看md5 确实是跟前面的还不一样的,说明这次是有改进的 :
0bf310d02e4d00b521a563a9d2c5dde8 patch/Sample1_L1_1.fq.gz
28b8c43a0640a1f6c3eaf2b60b1a4879 patch/Sample7_L2_2.fq.gz
cf1cff6f03dab2ebf87b32c8fe30e4c6 patch/Sample7_L4_1.fq.gz
1777b6d89c6dd4a3f209c4f66580bd95 patch/Sample7_L4_2.fq.gz
然后针对失败的两个样品,重新跑上面的脚本文本文件( run_sapceranger.sh ),发现Sample7跑流程仍然是失败的,说明还是有文件不完整,继续进行 gzip -t 的检验,发现是这个Sample7_S4_L001_R2_001.fastq.gz文件还是有问题,反反复复下载了四次,才最后通过了检验。
全部的6个样品跑完了之后,就可以整理他们的输出文件,然后去R里面做后续的分析咯:
pro_folder=outputs
mkdir -p $pro_folder
ls -lh $pro_folder
mkdir -p $pro_folder/html
ls */outs/web_summary.html |while read id;do (cp $id $pro_folder/html/${id%%/*}.html );done
mkdir -p $pro_folder/matrix
ls -d */outs/spatial |while read id;do (mkdir -p $pro_folder/matrix/${id%%/*}/; cp -r $id $pro_folder/matrix/${id%%/*}/; cp ${id%%/*}/outs/filtered_feature_bc_matrix.h5 $pro_folder/matrix/${id%%/*}/ );done
但是文章里面写的是这6个样品,合起来是有两万多个spots,如下所示:

我看了看,自己跑出来的结果非常诡异。。。
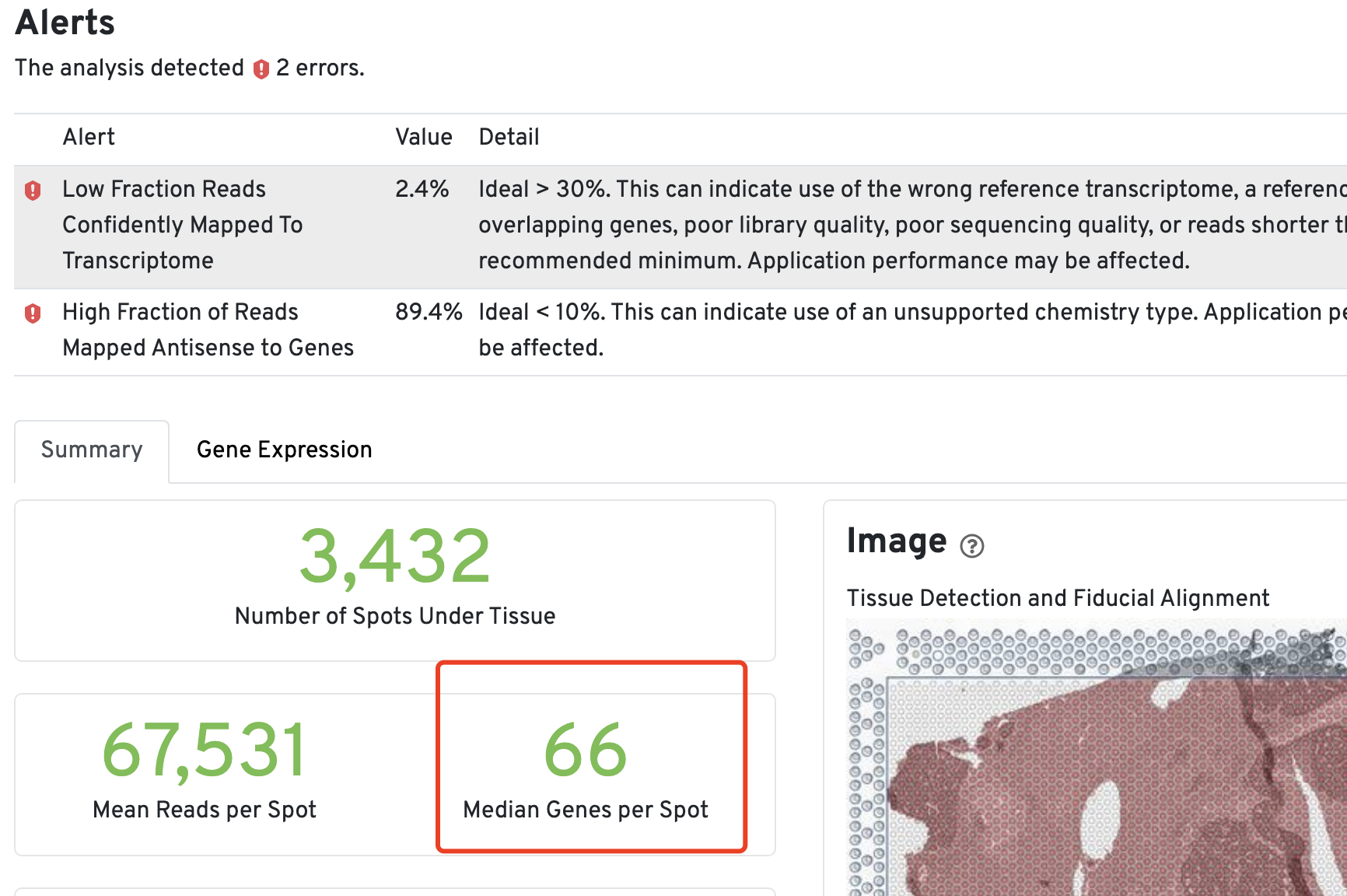
从这两个指标也能看出来诡异的地方就是这些测序片段都是可以比对到参考基因组,而且在基因坐标里面的,都是在外显子上面,非常合格的。就是不知道为什么都是Antisense ,不知道是不是软件有什么特殊的参数可以设置:
- Reads Mapped Confidently to Exonic Regions 96.8%
- Reads Mapped Antisense to Gene 91.7%