前面我们视频号直播了一个表达量芯片数据处理,详见:这样去除表达量芯片的批次效应可能不妥,这个物信息学数据挖掘的标题是:《Novel ferroptosis gene biomarkers and immune infiltration profiles in diabetic kidney disease via bioinformatics》,直播回放的视频在:
如果是常规的geo表达量芯片数据集代码,比如illumina的芯片,我们汇总了系列代码 :
https://www.jianguoyun.com/p/DdqkaeUQ1pC6BhixiLAFIAA
表达量芯片是非常适合锻炼大家的r编程基础的,新的一年从这3个gse数据集开始吧:
2015-GSE67936-AML-illumina
2016-GSE65409-AML-illumina
2019-GSE114868-AML-hta2.0
而且绝大部分表达量芯片并不需要从原始数据开始,比如affymetrix的芯片,一般来说就是读取作者给出来的 表达量矩阵文件即可,比如 GSE30122_series_matrix.txt.gz 文件是 7.0M ,可以看到它在线链接是有规律的:https://ftp.ncbi.nlm.nih.gov/geo/series/GSE30nnn/GSE30122/matrix/GSE30122_series_matrix.txt.gz
读取作者给出来的 表达量矩阵文件的标准代码如下所示:
library(AnnoProbe)
library(GEOquery)
getOption('timeout')
options(timeout=10000)
list.files()
if(T){gset <- geoChina(gse_number)}
if(F){ gset = getGEO("GSE30122", destdir = '.', getGPL = F) }
a=gset[[1]]
dat=exprs(a) #a现在是一个对象,取a这个对象通过看说明书知道要用exprs这个函数
dim(dat)#看一下dat这个矩阵的维度
dat[1:4,1:4] #查看dat这个矩阵的1至4行和1至4列,逗号前为行,逗号后为列
pd = pData(a)
head(pd)
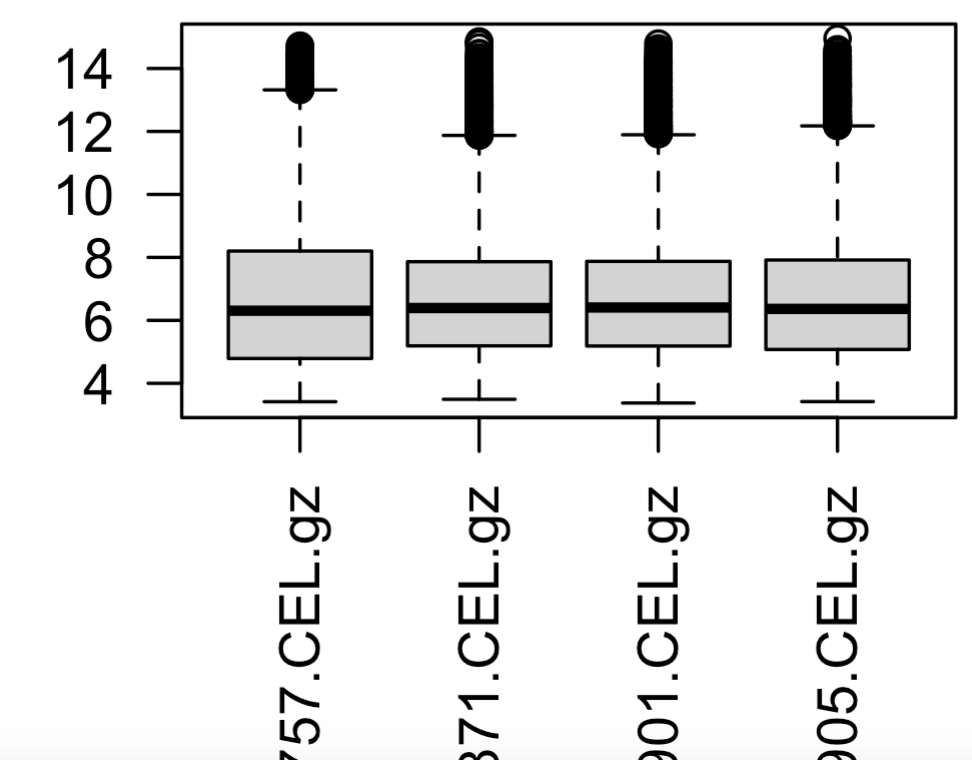
boxplot(dat[,1:4],las=2)
可以很明显的看到作者给出来的表达量矩阵里面的表达量是被zscore的 :
这个时候就需要下载这个项目的raw文件了,因为是affymetrix芯片,所以绝大部分是cel格式的文件 ,在线链接仍然是有规律的 :https://ftp.ncbi.nlm.nih.gov/geo/series/GSE30nnn/GSE30122/suppl/GSE30122_RAW.tar
同样的,很简单的读取它:
library(oligo)
celFiles <- list.celfiles('../cel_files/',listGzipped = T,
full.name=TRUE)
celFiles
options(BioC_mirror="https://mirrors.westlake.edu.cn/bioconductor")
exon_data <- oligo::read.celfiles( celFiles )
dim(exprs(exon_data))
exprs(exon_data)[1:4,1:4]
### 标准化(一步完成背景校正、分位数校正标准化和RMA (robust multichip average) 表达整合)
exon_data_rma <- oligo::rma(exon_data )
exp_probe <- Biobase::exprs(exon_data_rma)
exp_probe[1:4,1:4]
dat=exp_probe
dim(dat)#看一下dat这个矩阵的维度
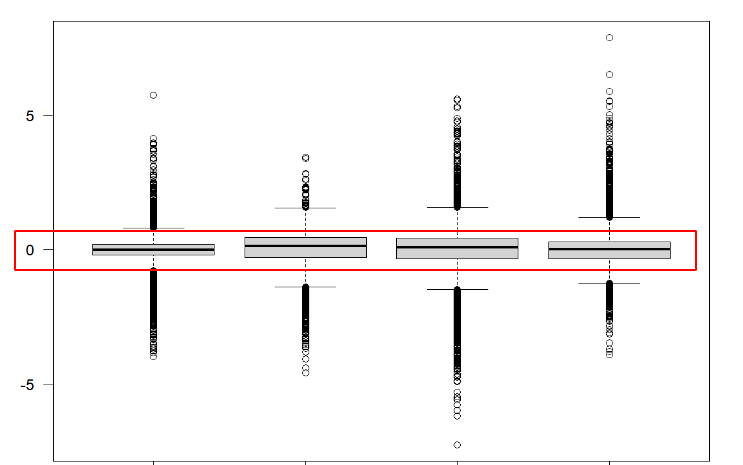
dat[1:4,1:4] #查看dat这个矩阵的1至4行和1至4列,逗号前为行,逗号后为列
如下所示看起来就很正常的表达量矩阵分布范围啦: