前面我们提到了目前绝大部分单细胞转录组数据分析相关文章都不约而同的使用了monocle2这个软件来做拟时序分析,但是并不意味着它是金标准,也不意味着非monocle2不可。
这个纯粹就是生物信息学领域的“马太效应”,大家都用monocle2做拟时序,所以后来者就简单的追随即可,而且绝大部分人其实并不关心算法细节,仅仅是为了做拟时序而做,那么就无所谓选择哪个软件了。我们也简单的展示了目前的可以做拟时序分析的软件的测评,详见:拟时序的多种算法大比拼(拟时序一本通03) 。但是,测评归测评,最终大家还是得使用monocle2做拟时序分析,所以不得不把重点放它的细节剖析上面,我们后面也会介绍一下其它软件和方法:
- monocle3拟时序实战
- SCORPIUS
- Slingshot
- 基于其它编程语言的拟时序分析
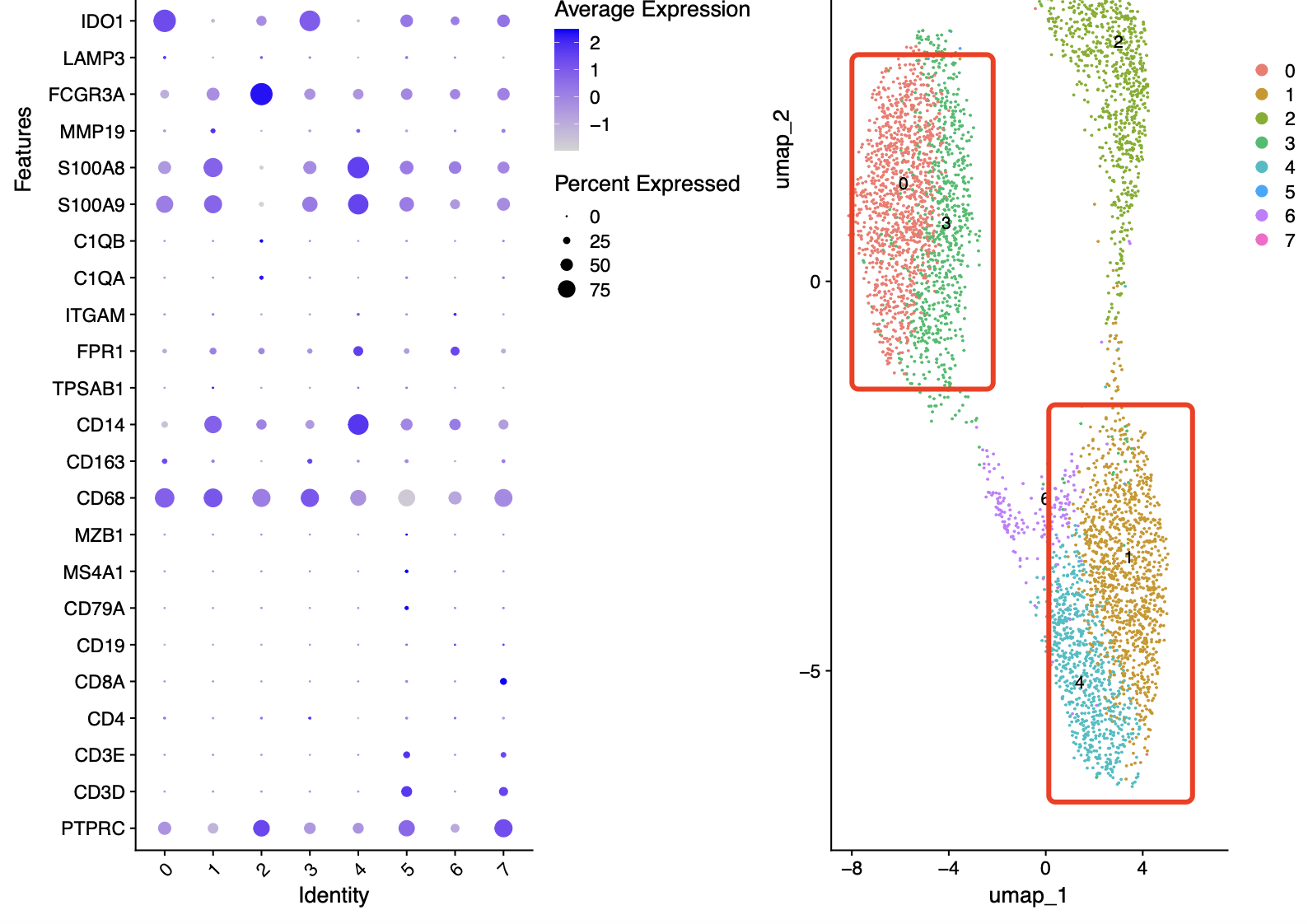
我们这里还是使用最开始的测试数据来作为案例演练,我们主要是关心的是已知的CD14和CD16的单核细胞的拟时序数据分析情况。首先大家需要有生物学背景知识,就是CD14和CD16是用于表征单核细胞(单核吞噬细胞)亚型的两种常见标记物。
首先看看单细胞转录组数据集情况:
这里我们使用的仍然是最经典的数据集,是来自于SeuratData包的ifnb数据集,详见;SCTransform真的能完美替代Seurat早期的3个函数吗。也是从这个数据集里面拿到CD14和CD16的单核细胞后进行后面的分析:
rm(list = ls())
library(Seurat)
library(SeuratData)
library(ifnb.SeuratData)
data("ifnb")
ifnb=UpdateSeuratObject(ifnb)
sce.all <- ifnb
sce.all$celltype = sce.all$seurat_annotations
sel.clust = "celltype"
scRNA <- SetIdent(sce.all, value = sel.clust)
table(scRNA@active.ident)
kp = scRNA$celltype %in% c('CD14 Mono' , 'CD16 Mono')
scRNA=scRNA[,kp]
scRNA$celltype=as.character(scRNA$celltype)
table(scRNA$celltype)
细分亚群
因为前面我们针对CD14和CD16的单核细胞取子集了,所以有必要进行细分亚群的操作,代码可以看 链接: https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh ,只需要是针对上面的scRNA变量进行操作即可,可以看到我们单核细胞子集里面仍然是有淋巴细胞的干扰 ,如下所示:
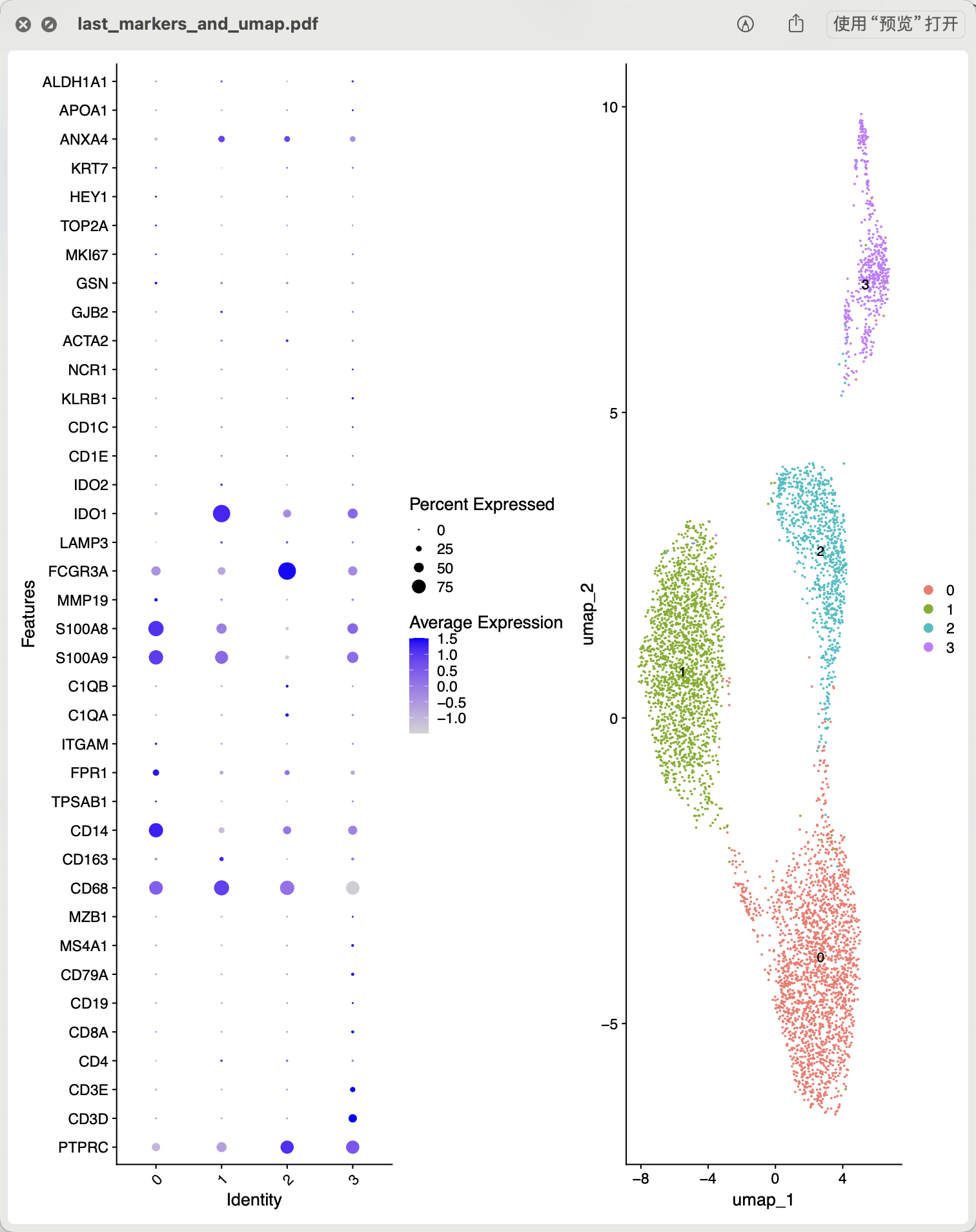
而且很有意思的是CD14单核细胞其实是有两个亚群的,而CD16就比较纯粹啦 :
cluster0:IL8,CLEC5A,IER3,S100A8,MARCKSL1,CD14
cluster1:CCL8,IDO1,RSAD2,IL1RN,CXCL11,APOBEC3A
cluster2:VMO1,MS4A4A,FCGR3A,PLAC8,HN1,PPM1N
cluster3:CD3D,CCR7,CD2,CD7,OCIAD2,LTB
而且很有意思的其实CD14单核细胞的两个亚群就相当于是来源于两个不同的样品:
IMMUNE_CTRL IMMUNE_STIM
0 1987 70
1 26 1866
2 431 480
3 274 265
这样的话,就很难分清楚具体是不同的CD14单核细胞亚群呢,还是CD14单核细胞的差异变化。
然后就是虽然是这个monocle2包可以做降维聚类分群,但是因为我们已经是使用了Seurat就做了上面的降维聚类分群,并且可视化给大家看了,所以不建议大家使用monocle2包本身的降维聚类分群了哈,使用Seurat的降维聚类分群代码可以看 链接: https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh ,只需要是针对上面的scRNA变量进行操作即可。
创造一个函数包装monocle2的拟时序分析
前面在为什么做拟时序 (展示差异细节)我们简单的演示了如何做一个极简的使用monocle2包的拟时序分析 ,就是构建好对象后的挑选基因,降维,排序这3个步骤即可,所以我们把它封装成为了一个函数,run_monocle2,如下所示的内容:
run_monocle2 <- function(seurat,outfile,rm=T){
#outfile='cds_by_RNA_snn_res.0.1.Rdata'
library(monocle)
expr_matrix=seurat@assays$RNA$counts#使用counts表达值
sample_sheet<-seurat@meta.data#将实验信息赋值新变量
gene_annotation=data.frame(gene_short_name=rownames(seurat))#构建一个含有基因名字的数据框
rownames(gene_annotation)=rownames(seurat)#将上述数据框的行名赋值基因名字
pd <- new("AnnotatedDataFrame", data = sample_sheet)#将实验信息变量转化为monocel可以接收的对象
fd <- new("AnnotatedDataFrame", data = gene_annotation)#将基因注释变量转化为monocle可以接收的对象
cds <- newCellDataSet(expr_matrix, phenoData = pd, featureData = fd,
expressionFamily=negbinomial.size())#创建一个monocle的对象
cds #cellData对象;monocle独有
cds <- estimateSizeFactors(cds)
cds <- estimateDispersions(cds)
## 拟时序只需要3个步骤:
# 第一步: 挑选合适的基因. 有多个方法,
# 例如提供已知的基因集,或者自己做差异分析后拿到的统计学显著的差异基因列表
if(rm){
diff_test_res <- differentialGeneTest(cds,
fullModelFormulaStr = " ~ celltype ",
reducedModelFormulaStr = " ~ orig.ident",
relative_expr=TRUE, cores=4)
}else{
diff_test_res <- differentialGeneTest(cds,
fullModelFormulaStr = " ~ celltype ",
#reducedModelFormulaStr = " ~ orig.ident",
relative_expr=TRUE, cores=4)
}
ordering_genes <- row.names (subset(diff_test_res, qval < 0.01))
ordering_genes
cds <- setOrderingFilter(cds, ordering_genes)
plot_ordering_genes(cds)
# 第二步: 降维。降维的目的是为了更好的展示数据。函数里提供了很多种方法,
# 不同方法的最后展示的图都不太一样, 其中“DDRTree”是Monocle2使用的默认方法
if(rm){
cds <- reduceDimension(cds,
max_components = 2,
num_dim = 6,
reduction_method = 'DDRTree',
residualModelFormulaStr = "~orig.ident", #去除样本影响
verbose = F)
}
else{
cds <- reduceDimension(cds,
max_components = 2,
num_dim = 6,
reduction_method = 'DDRTree',
#residualModelFormulaStr = "~orig.ident", #去除样本影响
verbose = F)
}
# 第三步: 对细胞进行排序
cds <- orderCells(cds)
save(cds,file = outfile)
}
这个run_monocle2函数可以选择是否抹除个体差异,这个很重要,而且它对拟时序的结果影响会很大!让我们再次回顾一下这个数据集,其中CD14单核细胞的两个亚群就相当于是来源于两个不同的样品:
IMMUNE_CTRL IMMUNE_STIM
0 1987 70
1 26 1866
2 431 480
3 274 265
也就是说0和1都是CD14单核细胞,而2群是CD16单核细胞,最后的3群是淋巴细胞。
全部单细胞亚群的拟时序
使用上面的run_monocle2函数即可,如下所示:
source('scRNA_scripts/lib.R')
seurat = readRDS('2-harmony/sce.all_int.rds')
seurat$celltype=seurat$RNA_snn_res.0.1
run_monocle2(seurat,'cds_by_all.Rdata')
library(monocle)
library(ggplot2)
library(patchwork)
load('cds_by_all.Rdata')
plot_cell_trajectory(cds, color_by = "orig.ident") +
plot_cell_trajectory(cds, color_by = "celltype")
ggsave(filename = 'celltype_vs_orig.ident_by_all.pdf',width = 10)
值得注意的是,针对全部单细胞亚群的拟时序是不可取的,因为这里面有3群是淋巴细胞,它理论上是跟另外的CD14单核细胞和CD16单核细胞没有发育关系,但是并不影响我们跑代码啦:
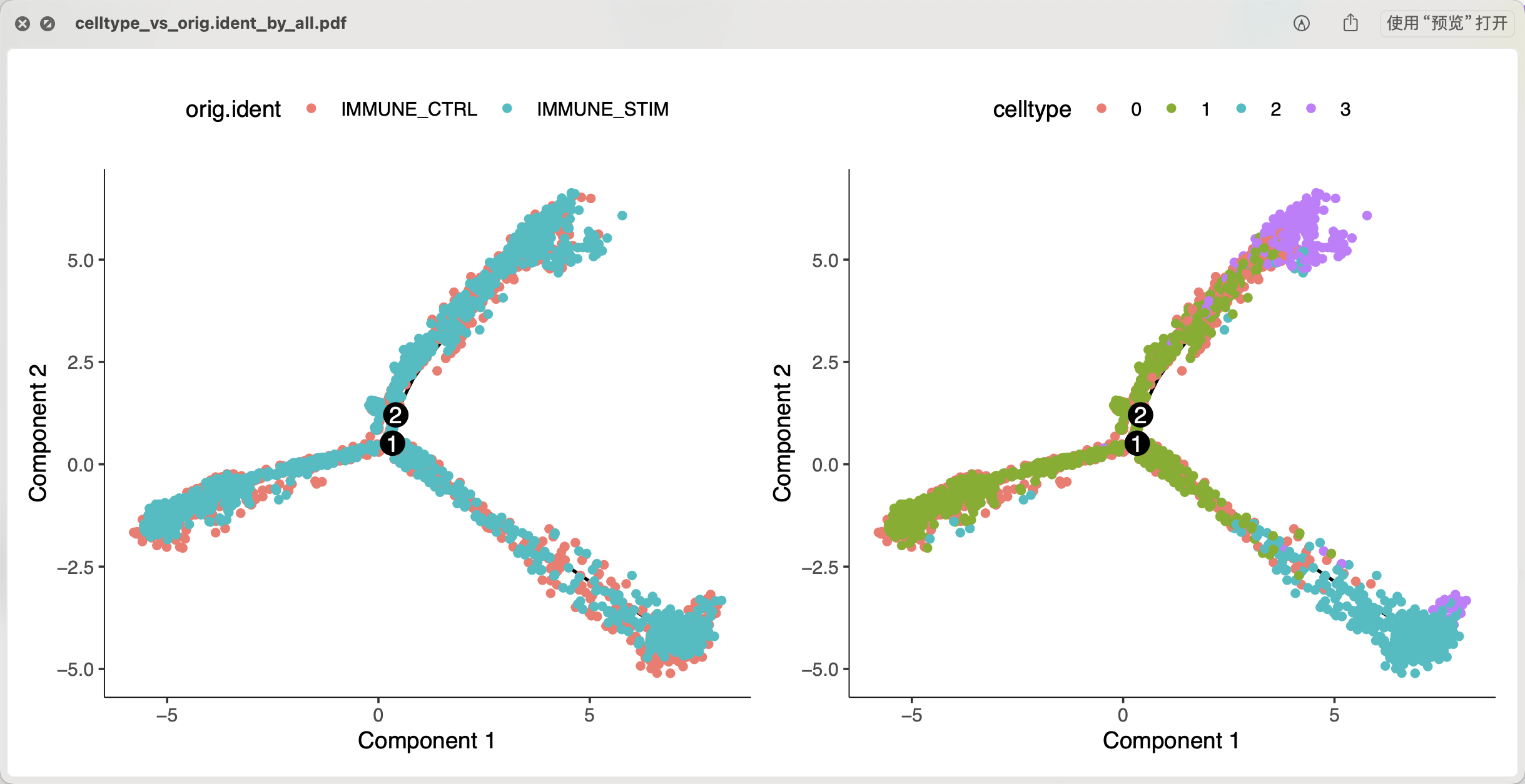
但是如果没有这样的生物学背景,就 很容易得到结论是CD14单核细胞会有两个发育方向,一个是我们熟知的CD16单核细胞,另外一个发育方向是CD14单核细胞演变成为了淋巴细胞。。。
去除淋巴细胞之后的拟时序
从生物学背景的角度,CD14单核细胞演变成为了淋巴细胞是不太可能的,所以我们应该是纯粹关心我们的CD14单核细胞和CD16单核细胞发育关系,如下所示:
seurat=seurat[,seurat$celltype %in% c(0,1,2)]
run_monocle2(seurat,'cds_by_remove_Tcells.Rdata')
load('cds_by_remove_Tcells.Rdata')
plot_cell_trajectory(cds, color_by = "orig.ident") +
plot_cell_trajectory(cds, color_by = "celltype")
ggsave(filename = 'celltype_vs_orig.ident_by_remove_Tcells.pdf',width = 10)
可以看到,这个时候我们的CD14单核细胞有两个截然不同的亚群, 它们都有可能演变成为CD16单核细胞。当然了,这个发育是没有方向的,所以理论上也可以说是CD16单核细胞会演变成为两个截然不同的CD14单核细胞啦。
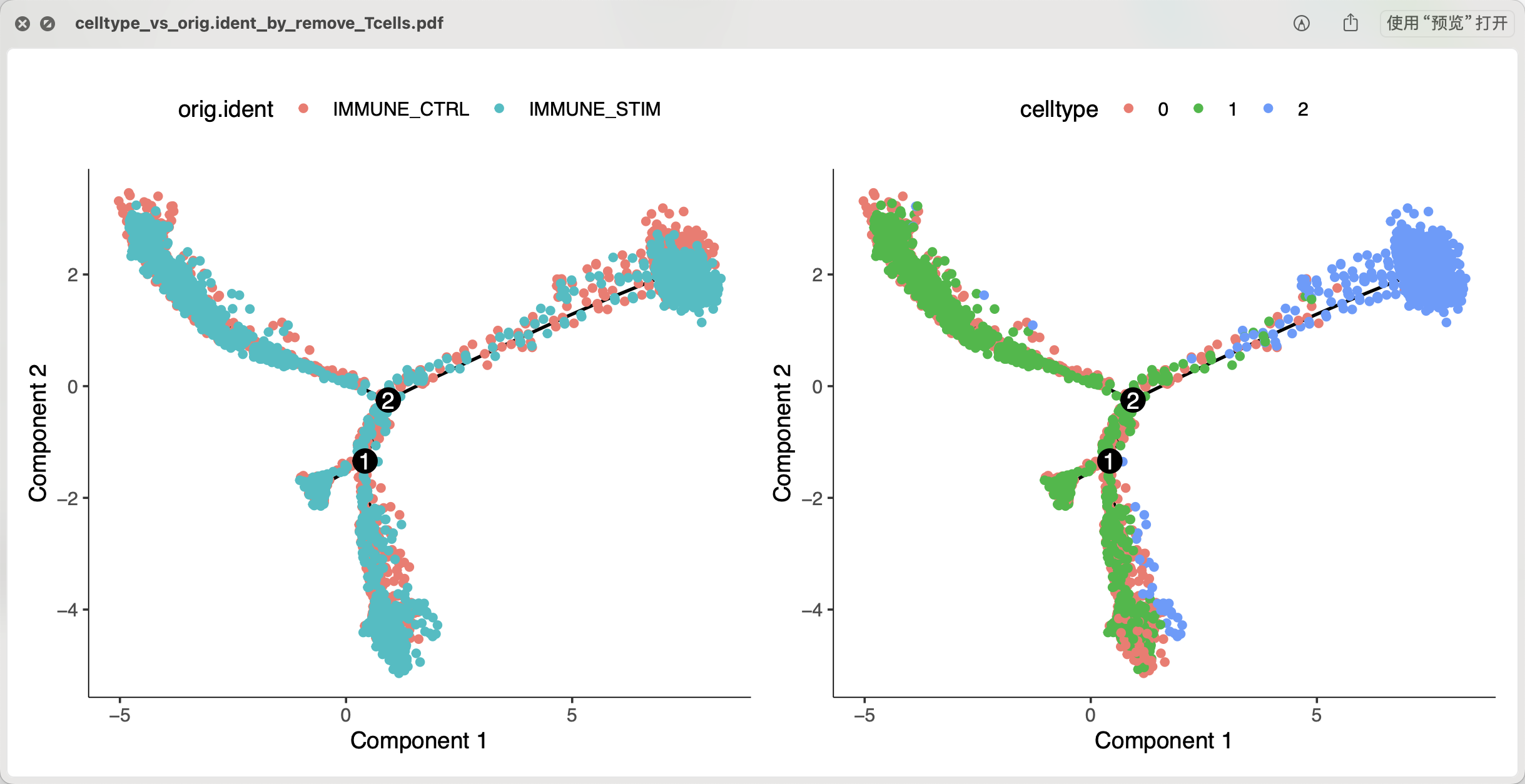
看第一群CD14单核细胞和CD16单核细胞拟时序关系
seurat = readRDS('2-harmony/sce.all_int.rds')
seurat$celltype=seurat$RNA_snn_res.0.1
seurat=seurat[,seurat$celltype %in% c(0,2 )]
run_monocle2(seurat,'cds_by_0_and_2.Rdata')
load('cds_by_0_and_2.Rdata')
plot_cell_trajectory(cds, color_by = "orig.ident") +
plot_cell_trajectory(cds, color_by = "celltype")
ggsave(filename = 'celltype_vs_orig.ident_by_0_and_2.pdf',width = 10)
因为这个时候的0这个编号亚群的CD14单核细胞基本上就是属于IMMUNE_CTRL样品的,所以如果我们这个时候抹去个体差异后做拟时序,结果会很诡异,如下所示:
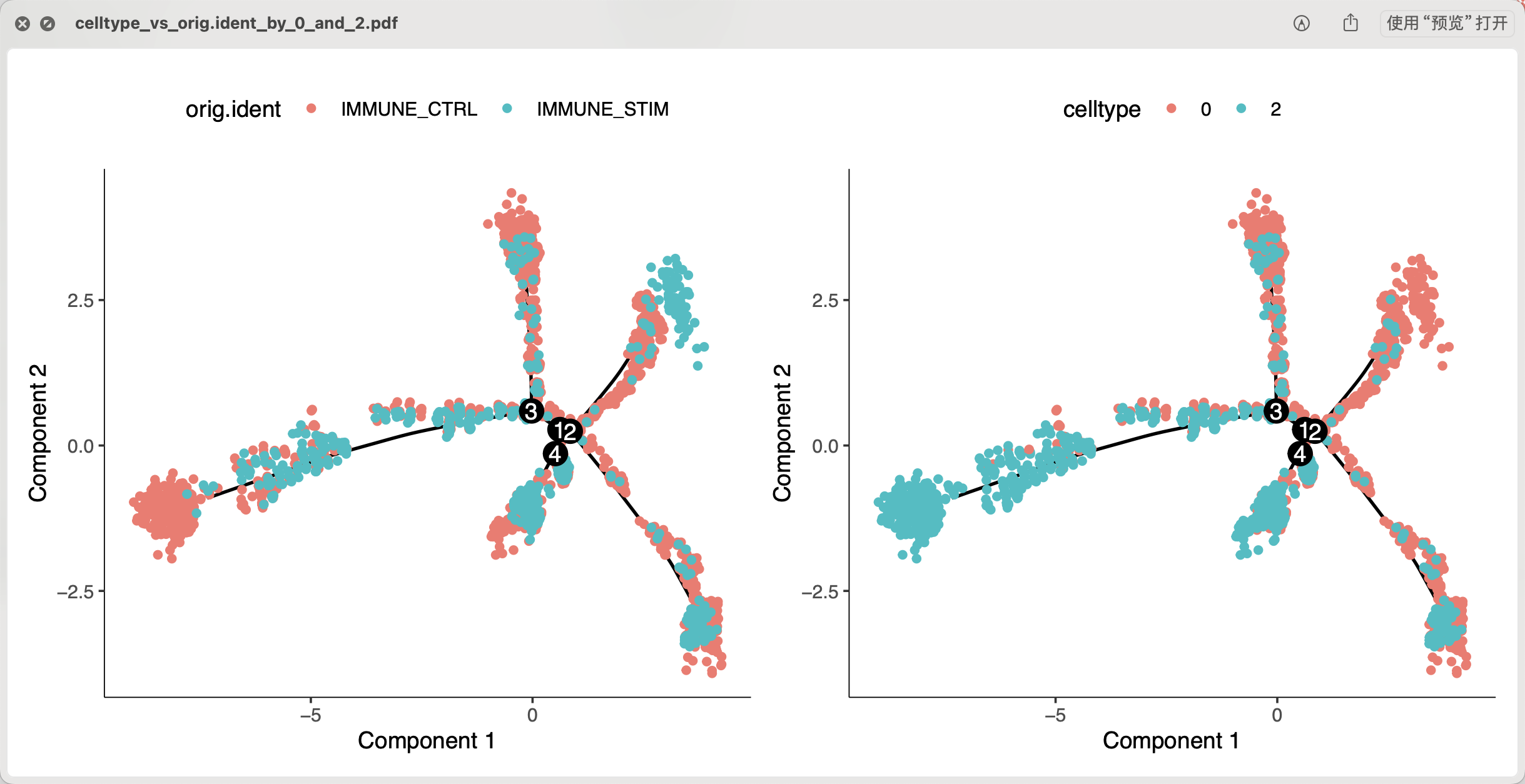
结果非常不好解释,也不符合我们的预期,因为我们提取了0这个编号亚群的CD14单核细胞理论上它比较纯粹了,它去跟CD16单核细胞拟时序理论上就应该是线性变化,所以我们试试看不要抹去个体差异后做拟时序,因为这个时候的个体差异本身也是两个亚群的差异:
seurat = readRDS('2-harmony/sce.all_int.rds')
seurat$celltype=seurat$RNA_snn_res.0.1
seurat=seurat[,seurat$celltype %in% c(0,2 )]
run_monocle2(seurat,'cds_by_0_and_2_not_rm.Rdata',rm = F)
load('cds_by_0_and_2_not_rm.Rdata')
plot_cell_trajectory(cds, color_by = "orig.ident") +
plot_cell_trajectory(cds, color_by = "celltype")
ggsave(filename = 'celltype_vs_orig.ident_by_0_and_2_not_rm.pdf',width = 10)
可以看到,如下所示不抹去个体差异后做拟时序,是可以比较清晰我们实际上做的并不是我们感兴趣的两个单细胞亚群的演变关系(差异细节),而是纯粹的两个个体的演变关系(差异细节),也未必是我们的想要的结果啦 :
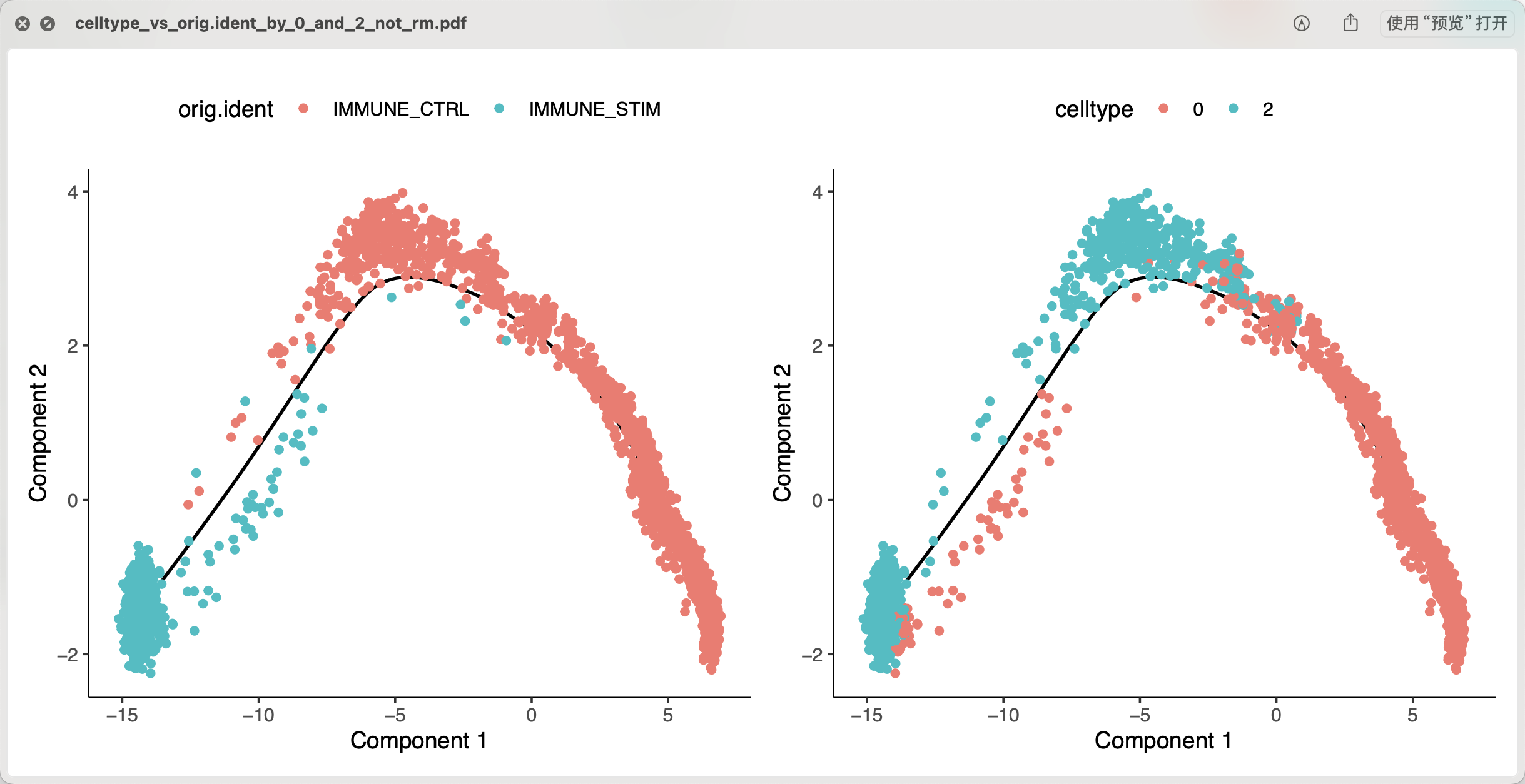
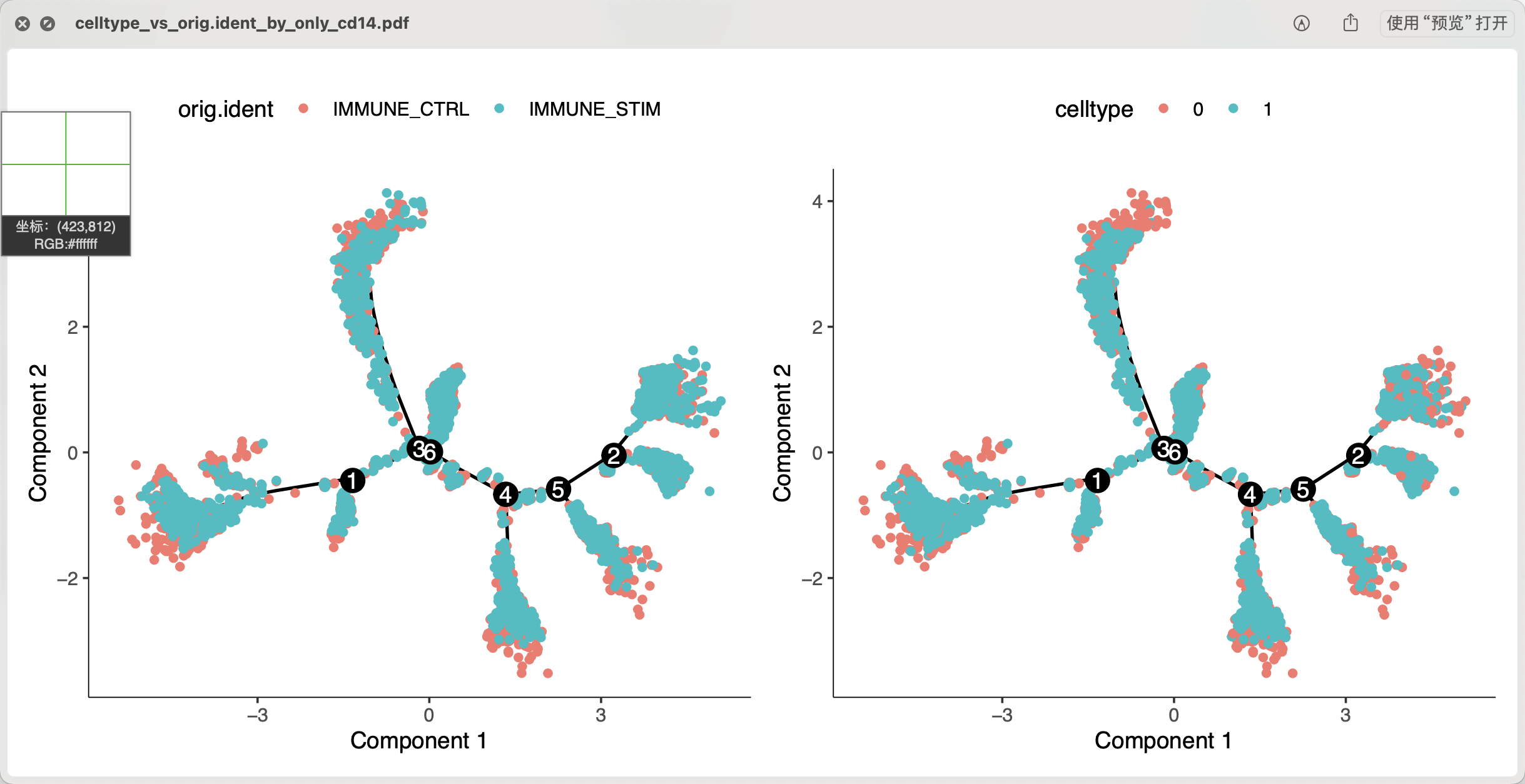
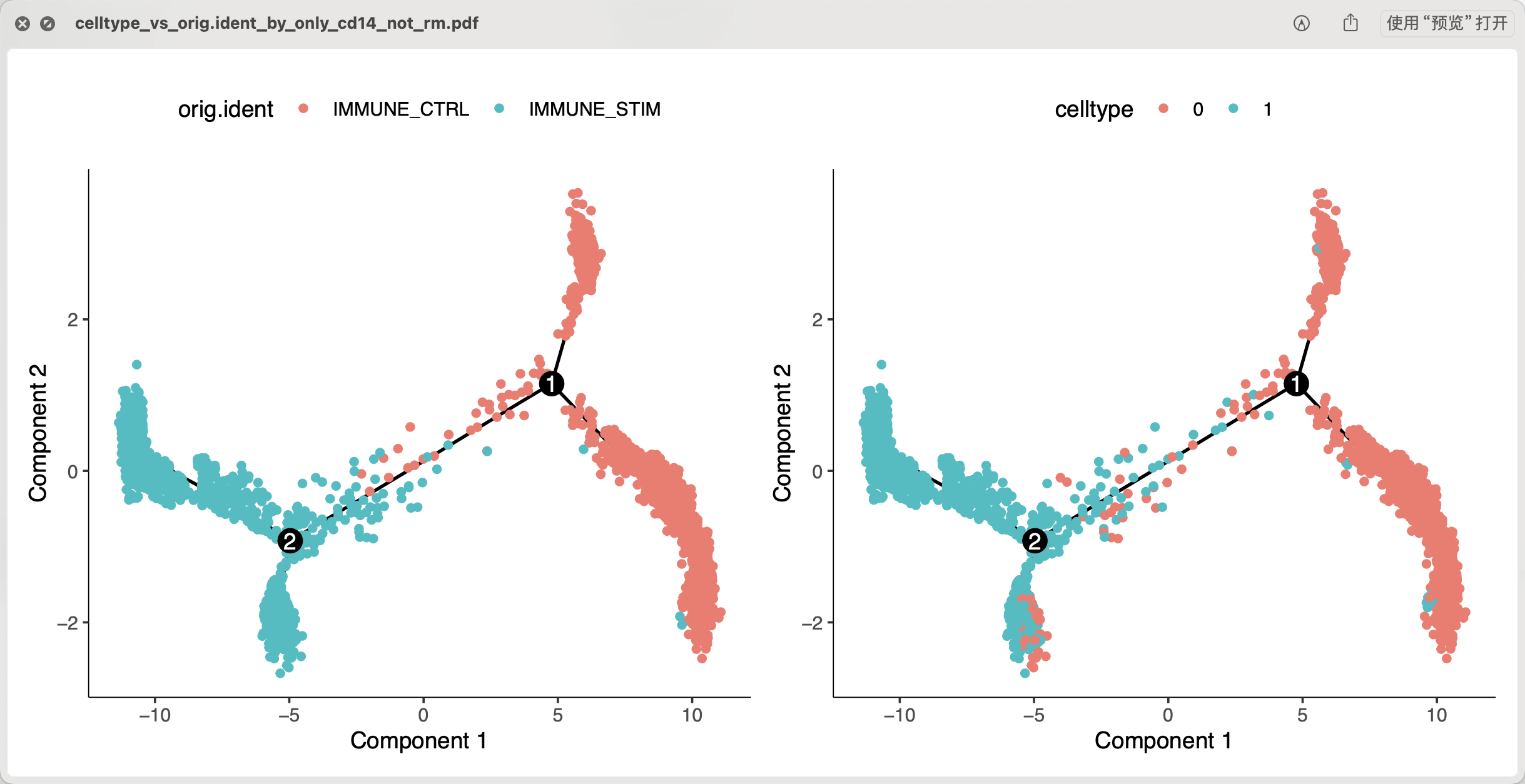
如果我们把前面的0这个编号亚群的CD14单核细胞替换成1这个编号亚群,做同样的分析,可以看到类似的结果:
上面的是抹去个体差异后做拟时序,下面的不抹去个体差异后做拟时序,可以看到基本上也是类似的情况。
如果仅仅是看两个不同编号的CD14单核细胞的关系
因为这个0和1的编号亚群其实就是就相当于是来源于两个不同的样品:
IMMUNE_CTRL IMMUNE_STIM
0 1987 70
1 26 1866
2 431 480
3 274 265
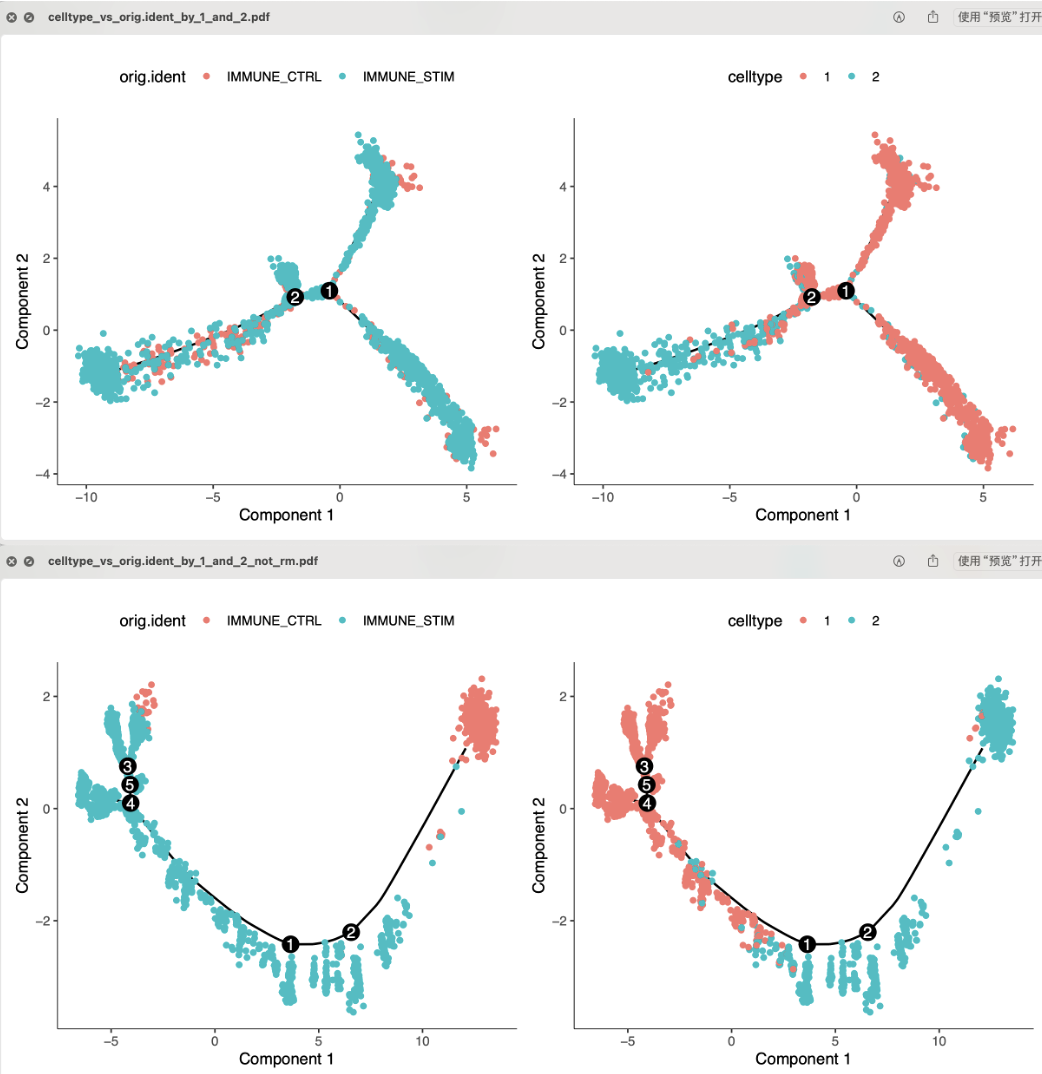
也就是说0和1都是CD14单核细胞,而2群是CD16单核细胞,最后的3群是淋巴细胞。所以如果我们仅仅是拿0和1的编号亚群CD14单核细胞去做拟时序,这个时候我们就算是抹去个体差异后做拟时序,结果也会很诡异,像一个蜈蚣一样的,如下所示:
其实之前我们就摸索过,详见:听说你的拟时序图跑的像蜈蚣,那个时候是因为没有抹去个体差异后做拟时序所以结果很多分叉,但是这个时候是因为抹去个体差异后做拟时序结果很多分叉,所以我也尝试一下不抹除个体差异后做拟时序,确实是有改变,如下所示:
实际上,这个时候我们并没有标准答案,因为这个0和1的编号CD14单核细胞亚群其实就是就相当于是来源于两个不同的样品,如果抹除样品差异那么就根本就看不到两个不同子亚群了所以做拟时序结果很多分叉,如果不抹除样品差异那么确实是有两个来源于不同样品的亚群,但是拟时序展现的差异细节仍然是会被个体差异掩盖掉。
上面的3种不同选择的拟时序过程的代码是:
{
seurat=seurat[,seurat$celltype %in% c(0,1 )]
run_monocle2(seurat,'cds_by_only_cd14.Rdata')
seurat = readRDS('2-harmony/sce.all_int.rds')
seurat$celltype=seurat$RNA_snn_res.0.1
seurat=seurat[,seurat$celltype %in% c(0,2 )]
run_monocle2(seurat,'cds_by_0_and_2.Rdata')
seurat = readRDS('2-harmony/sce.all_int.rds')
seurat$celltype=seurat$RNA_snn_res.0.1
seurat=seurat[,seurat$celltype %in% c(1,2 )]
run_monocle2(seurat,'cds_by_1_and_2.Rdata')
}
{
seurat=seurat[,seurat$celltype %in% c(0,1 )]
run_monocle2(seurat,'cds_by_only_cd14_not_rm.Rdata',rm = F)
seurat = readRDS('2-harmony/sce.all_int.rds')
seurat$celltype=seurat$RNA_snn_res.0.1
seurat=seurat[,seurat$celltype %in% c(0,2 )]
run_monocle2(seurat,'cds_by_0_and_2_not_rm.Rdata',rm = F)
seurat = readRDS('2-harmony/sce.all_int.rds')
seurat$celltype=seurat$RNA_snn_res.0.1
seurat=seurat[,seurat$celltype %in% c(1,2 )]
run_monocle2(seurat,'cds_by_1_and_2_not_rm.Rdata',rm = F)
}
然后针对拟时序的结果的可视化代码是:
{
load('cds_by_only_cd14.Rdata')
plot_cell_trajectory(cds, color_by = "orig.ident") +
plot_cell_trajectory(cds, color_by = "celltype")
ggsave(filename = 'celltype_vs_orig.ident_by_only_cd14.pdf',width = 10)
load('cds_by_0_and_2.Rdata')
plot_cell_trajectory(cds, color_by = "orig.ident") +
plot_cell_trajectory(cds, color_by = "celltype")
ggsave(filename = 'celltype_vs_orig.ident_by_0_and_2.pdf',width = 10)
load('cds_by_1_and_2.Rdata')
plot_cell_trajectory(cds, color_by = "orig.ident") +
plot_cell_trajectory(cds, color_by = "celltype")
ggsave(filename = 'celltype_vs_orig.ident_by_1_and_2.pdf',width = 10)
}
{
load('cds_by_only_cd14_not_rm.Rdata')
plot_cell_trajectory(cds, color_by = "orig.ident") +
plot_cell_trajectory(cds, color_by = "celltype")
ggsave(filename = 'celltype_vs_orig.ident_by_only_cd14_not_rm.pdf',width = 10)
load('cds_by_0_and_2_not_rm.Rdata')
plot_cell_trajectory(cds, color_by = "orig.ident") +
plot_cell_trajectory(cds, color_by = "celltype")
ggsave(filename = 'celltype_vs_orig.ident_by_0_and_2_not_rm.pdf',width = 10)
load('cds_by_1_and_2_not_rm.Rdata')
plot_cell_trajectory(cds, color_by = "orig.ident") +
plot_cell_trajectory(cds, color_by = "celltype")
ggsave(filename = 'celltype_vs_orig.ident_by_1_and_2_not_rm.pdf',width = 10)
}
也就是说, 哪怕是看起来很简单的拟时序分析,仅仅是跑一次代码就可以拿到结果,也会面临其数据分析对象的选择问题以及分析过程的参数选择问题的纠结。
什么是合理的拟时序分析呢
如果我们继续提高降维聚类分群的分辨率,可以得到如下所示的结果:
> table(seurat$RNA_snn_res.0.8,seurat$orig.ident)
IMMUNE_CTRL IMMUNE_STIM
0 3 1140
1 1129 5
2 434 480
3 38 740
4 709 4
5 223 205
6 131 47
7 51 60
之前的分辨率低的时候,编号0和编号1都是CD14单核细胞,而编号2群是CD16单核细胞,最后的编号3群是淋巴细胞。现在分辨率高了,之前的编号0群就裂变成为了 现在的编号0和编号3亚群,而之前的编号1群就裂变成为了现在的编号1和编号4亚群,仍然是具有很明显的个体异质性。之前的2群是CD16单核细胞,现在的编号2群也是CD16单核细胞它也同样的分布在两个不同样品里面。
我们关心什么差异,就应该是做什么样的拟时序分析,需要牢记为什么做拟时序 (展示差异细节)。让我们再次回顾一下这个数据集在低分辨率情况下的降维聚类分群结果,其中CD14单核细胞的两个亚群就相当于是来源于两个不同的样品:
IMMUNE_CTRL IMMUNE_STIM
0 1987 70
1 26 1866
2 431 480
3 274 265
也就是说0和1都是CD14单核细胞,而2群是CD16单核细胞,最后的3群是淋巴细胞。也就是说可以有很多组合去做展示差异细节,比如我们还可以首先区分个体,然后再做拟时序,这个时候就无所谓是否抹除个体差异啦,因为本来就已经是在每个个体内部做拟时序了。代码如下所示:
{
seurat = readRDS('2-harmony/sce.all_int.rds')
seurat$celltype=seurat$RNA_snn_res.0.1
seurat=seurat[,seurat$celltype %in% c(0,2 )]
seurat=seurat[,seurat$orig.ident %in% c('IMMUNE_CTRL' )]
run_monocle2(seurat,'cds_by_0_and_2_IMMUNE_CTRL.Rdata',rm = F)
seurat = readRDS('2-harmony/sce.all_int.rds')
seurat$celltype=seurat$RNA_snn_res.0.1
seurat=seurat[,seurat$celltype %in% c(1,2 )]
seurat=seurat[,seurat$orig.ident %in% c('IMMUNE_STIM' )]
run_monocle2(seurat,'cds_by_1_and_2_IMMUNE_STIM.Rdata',rm = F)
load('cds_by_0_and_2_IMMUNE_CTRL.Rdata')
plot_cell_trajectory(cds, color_by = "orig.ident") +
plot_cell_trajectory(cds, color_by = "celltype")
ggsave(filename = 'celltype_vs_orig.ident_by_0_and_2_IMMUNE_CTRL.pdf',width = 10)
load('cds_by_1_and_2_IMMUNE_STIM.Rdata')
plot_cell_trajectory(cds, color_by = "orig.ident") +
plot_cell_trajectory(cds, color_by = "celltype")
ggsave(filename = 'celltype_vs_orig.ident_by_1_and_2_IMMUNE_CTRL.pdf',width = 10)
}
因为编号2群是CD16单核细胞,而编号0和1群是CD14单核细胞,所以可以看到的是在每个个体内部其实CD14单核细胞都是有子集的,结论是CD16单核细胞会演变成为两个截然不同的CD14单核细胞,反之亦然,因为拟时序分析没办法确定起点,就是揭示了这个变迁关系而已!
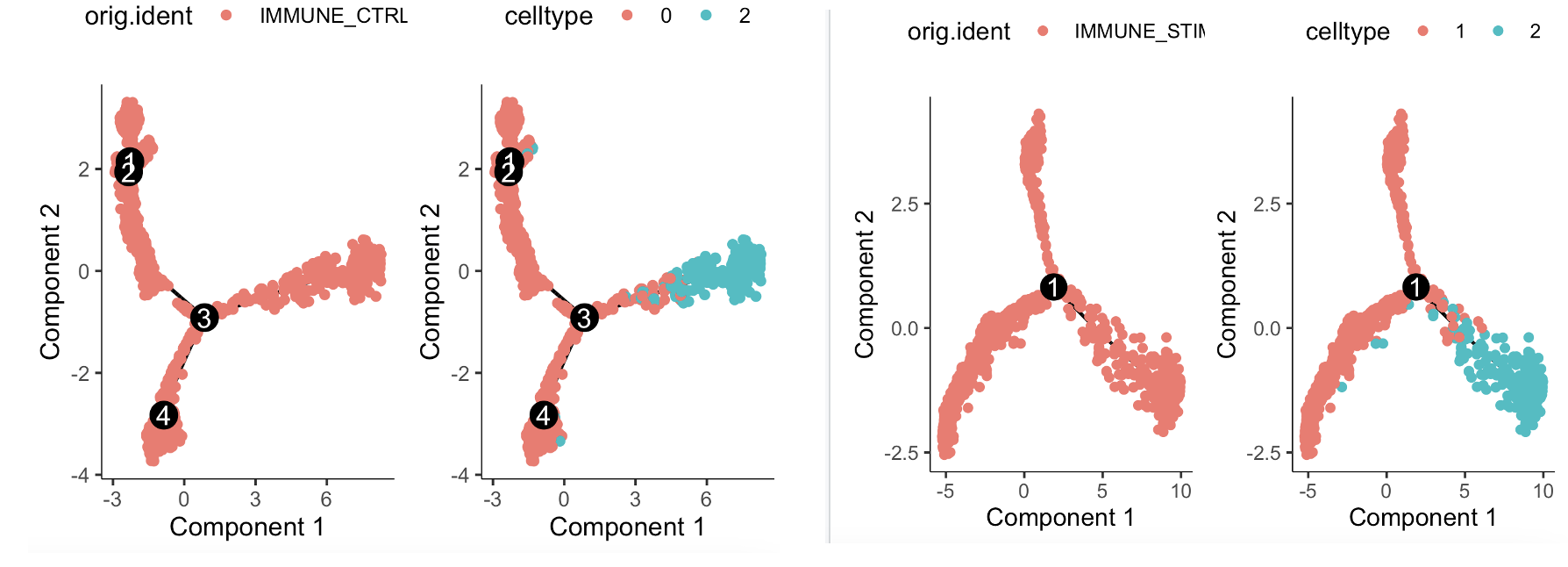
而且,这两个个体内部的独立的拟时序分析结果,是可以比较的。