去批次这个分析在我们组学数据分析领域非常常见, 可以提高数据质量,确保分析结果的准确性和可靠性。然而,去批次效应并不总是完美的,需要结合具体的数据特点和生物学背景,选择合适的方法,并进行仔细的验证和解释。
“去批次效应”(batch effect removal)这个步骤主要是:
- 实验设计:在进行大规模的组学实验时,由于成本、时间或技术限制,数据可能被分成多个批次进行收集。不同批次之间可能存在实验条件的微小变化,如操作人员、试剂批次、仪器状态等,这些因素都可能导致批次效应。
- 数据一致性:批次效应会导致数据在不同批次间产生系统性偏差,影响数据的一致性和可比性。如果不进行去批次效应处理,可能会对后续的数据分析和生物学解释产生误导。
- 统计分析:批次效应可能会掩盖或模糊化真实的生物学变异,使得统计分析的结果不准确。去除批次效应可以提高统计检验的可靠性。
- 机器学习:在利用机器学习算法进行模式识别或分类时,批次效应可能会引入噪声,影响模型的性能。去批次效应可以提高模型的泛化能力和预测准确性。
- 数据整合:在进行跨实验室或跨平台的数据整合时,去除批次效应可以提高数据的兼容性,使得来自不同来源的数据可以进行有效的比较和整合。
- 生物学解释:去除批次效应后,可以更准确地识别和解释生物学上有意义的变异,如基因表达的差异、蛋白质丰度的变化等。
- 研究复现性:去除批次效应有助于提高研究的复现性,因为去除了实验条件变化带来的影响,使得其他研究者可以在相似的条件下复现结果。
但是很多人理解的”去批次效应”(batch effect removal)这个操作应该是会输入一个表达量矩阵,然后输出一个表达量矩阵。其实在单细胞转录组数据分析里面并不是这样的,比如我们常见的harmony操作,它针对的就并不是原始的单细胞转录组表达量矩阵(几万个基因几万个细胞),而是pca分析结果(还是几万个细胞但是只有少量的pc)。这样的话,harmony操作后并没有修改我们的原始的单细胞转录组表达量矩阵,这一点可能会确实是让大家困惑。
DESeq2包本来就是可以把批次这个变量考虑进去
我们拿常规的转录组数据分析”去批次效应”(batch effect removal)这个操作举例来说明,详见:转录组测序的count矩阵如何去批次呢(sva包的ComBat_seq函数),这个案例里面就是会输入一个表达量矩阵,然后输出一个表达量矩阵。是大家比较容易理解的,但是如果我们是为了做转录组差异分析,其实是可以不需要输出一个表达量矩阵,因为DESeq2包本来就是可以把批次这个变量考虑进去,如下所示:
rm(list = ls())
load("Rdata/dat.Rdata")
exprSet[1:4,1:4]
table(group_list)
suppressMessages(library(DESeq2))
(colData <- data.frame(row.names=colnames(exprSet),
group_list=group_list,
batch= batch ) )
dds <- DESeqDataSetFromMatrix(countData = exprSet,
colData = colData,
design = ~ group_list+batch)
dds <- DESeq(dds)
resultsNames(dds)
res <- results(dds, name= resultsNames(dds)[2])
summary(res)
save(res,file = 'DESeq2_rm_batch.Rdata')
vsd <- vst(dds, blind = FALSE) #vsd 相当于标准化的 dds
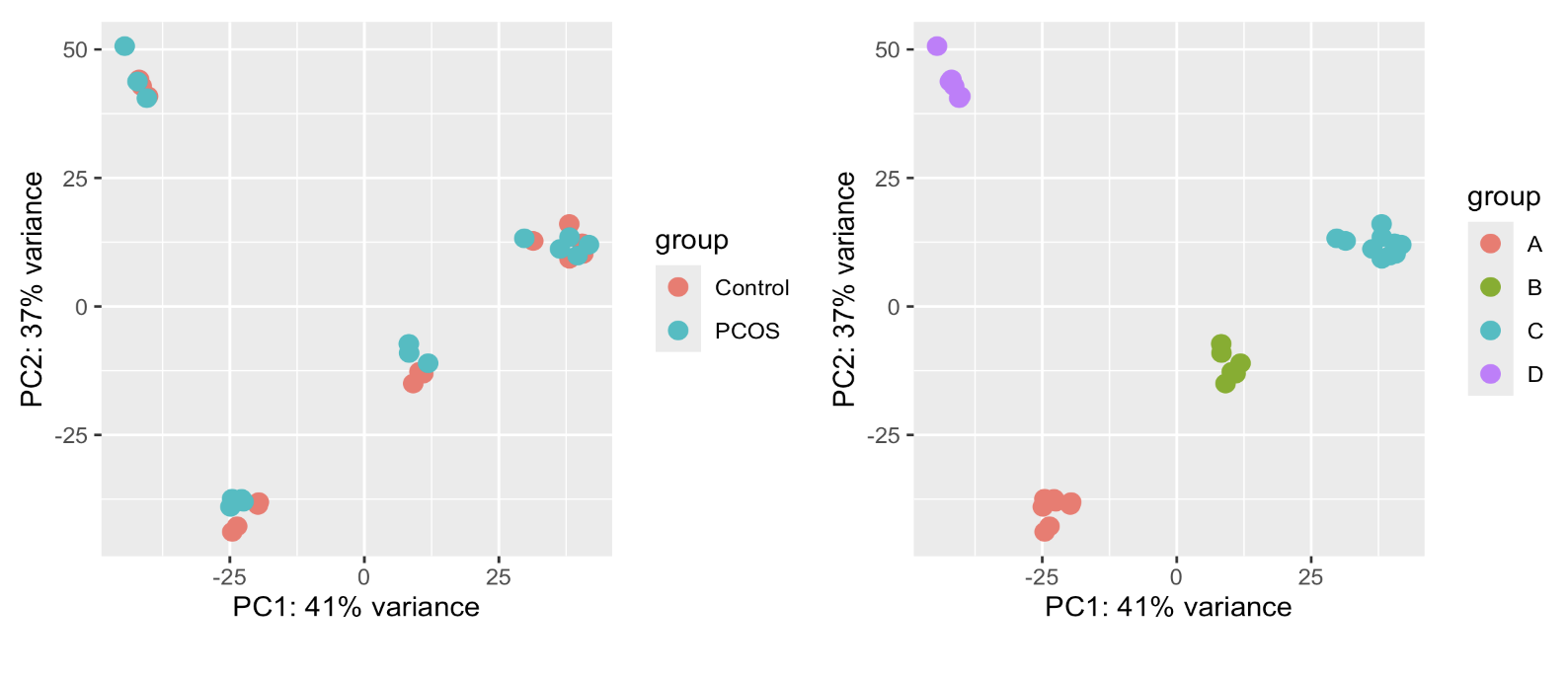
plotPCA(vsd, "group_list") + ##exprSet->dds->vsd
plotPCA(vsd, "batch")
可以看到,它针对原始的count表达量矩阵可以差异分析,但是如果我们可视化它的批次这个变量,可以看到其实并没有修改表达量矩阵,所以基于原始的表达量矩阵进行pca是看不到数据集的混合效应的。
如果我们是想修改表达量矩阵,还需要借助于其它包;
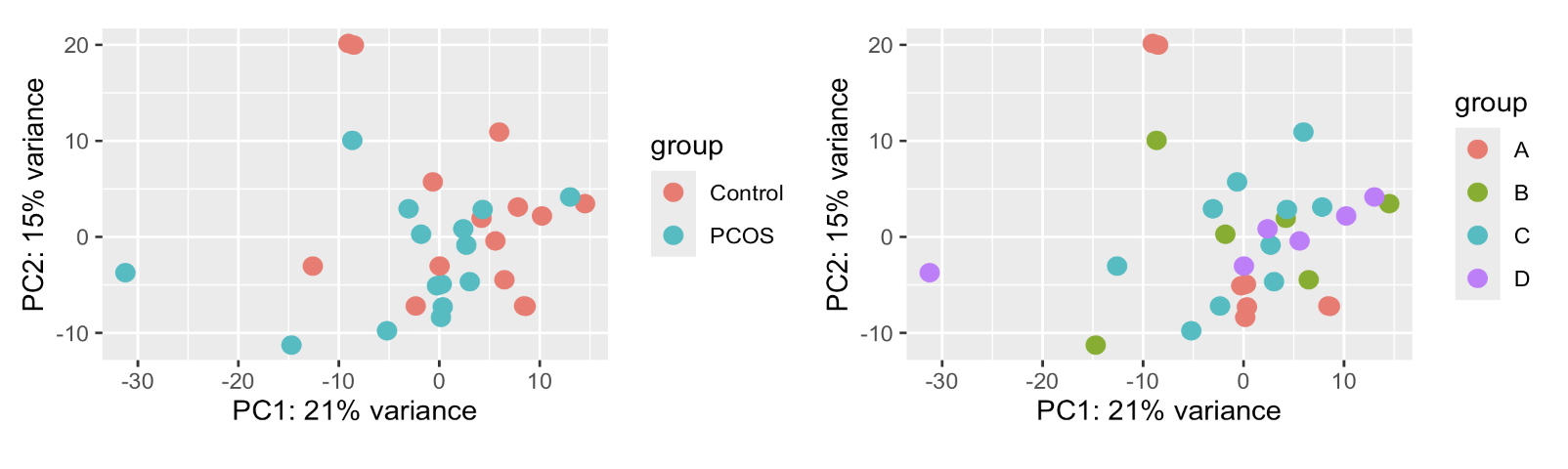
assay(vsd) <- limma::removeBatchEffect(assay(vsd), vsd$batch)
plotPCA(vsd, "group_list") + ##exprSet->dds->vsd
plotPCA(vsd, "batch")
如下所示,可以看到这个修改后的表达量矩阵其实就被抹除了批次这个变量造成的差异。关于这个数据集的介绍详见:转录组测序的count矩阵如何去批次呢(sva包的ComBat_seq函数)
两种不同的差异分析策略有什么区别呢
现在问题就来了,既然是可以不需要修改表达量矩阵,那么我们之前的操作详见:转录组测序的count矩阵如何去批次呢(sva包的ComBat_seq函数)又确实是拿到了修改后的表达量矩阵,基于它的差异分析结果跟我们的不修改表达量矩阵的差异分析是否有区别呢?
我们可以比较一下两次差异分析结果:
rm(list = ls())
load(file = 'DESeq2_rm_batch.Rdata')
summary(res)
deg_DESeq2 = na.omit(as.data.frame(res[order(res$padj),]))
load(file = 'DESeq2_rm_batch_combat.Rdata')
summary(res)
deg_combat = na.omit(as.data.frame(res[order(res$padj),]))
可以看到,如果不走sva包的ComBat_seq函数,也就是说不修改表达量矩阵,结果如下所示:
out of 15825 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 127, 0.8%
LFC < 0 (down) : 237, 1.5%
outliers [1] : 69, 0.44%
low counts [2] : 2132, 13%
(mean count < 3)
但是如果是sva包的ComBat_seq函数修改了表达量矩阵,结果是:
out of 15824 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 200, 1.3%
LFC < 0 (down) : 275, 1.7%
outliers [1] : 0, 0%
low counts [2] : 3683, 23%
(mean count < 6)
这样很难看出来区别,我们还是使用经典的散点图:
colnames(deg_combat)
ids=intersect(rownames(deg_DESeq2),
rownames(deg_combat))
df= data.frame(
deg_DESeq2 = deg_DESeq2[ids,'log2FoldChange'],
deg_combat = deg_combat[ids,'log2FoldChange']
)
library(ggpubr)
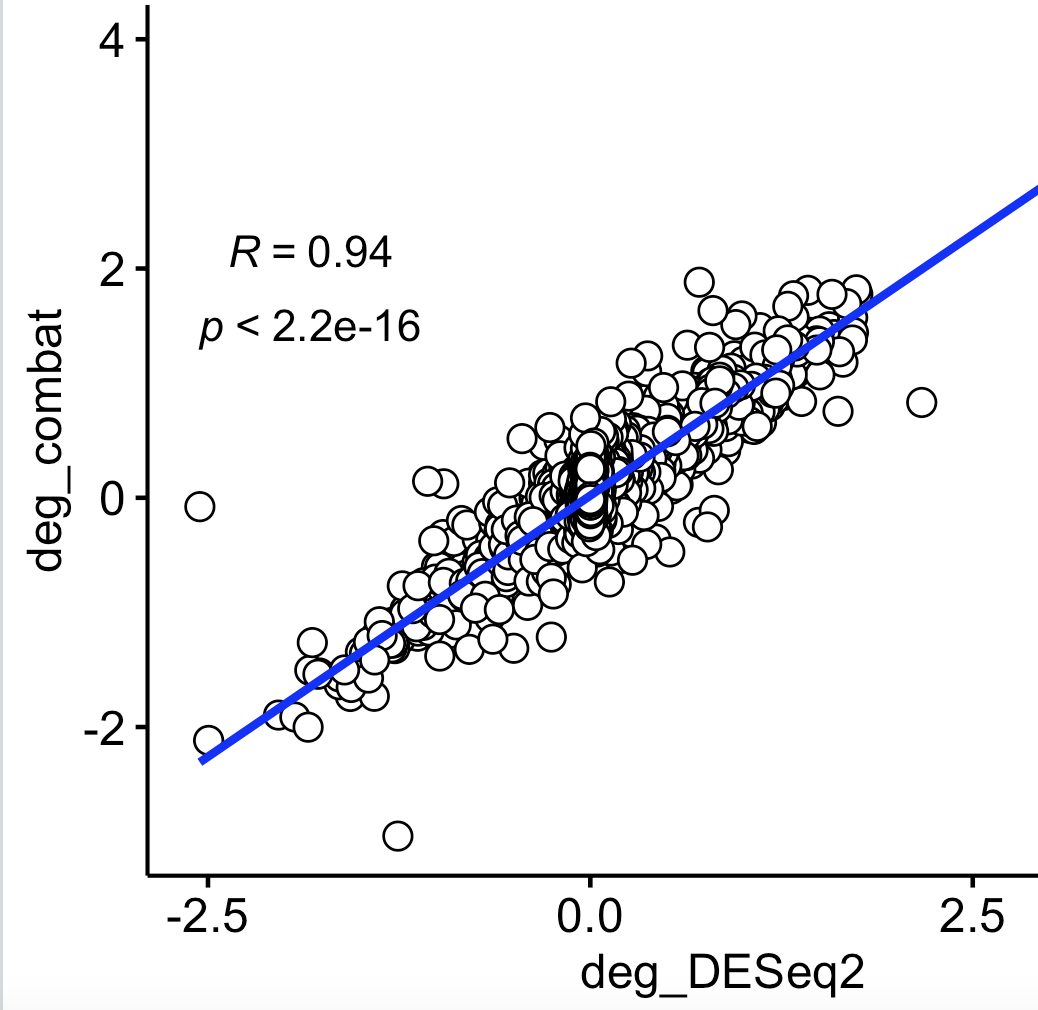
ggscatter(df, x = "deg_DESeq2", y = "deg_combat",
color = "black", shape = 21, size = 3, # Points color, shape and size
add = "reg.line", # Add regressin line
add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
conf.int = TRUE, # Add confidence interval
cor.coef = TRUE, # Add correlation coefficient. see ?stat_cor
cor.coeff.args = list(method = "pearson", label.sep = "\n")
)
可以看到,两种不同的差异分析策略其实并没有我们想象中的那么大的结果区别:
当然了,差异分析并不是仅仅是看log2FoldChange即可,还需要考虑统计学指标,比如p值和矫正的p值,我们也可以对比:
pvalue_t=0.05
logFC_t = 0.5
deg_DESeq2$g=ifelse(deg_DESeq2$padj > pvalue_t,'stable', #if 判断:如果这一基因的P.Value>0.01,则为stable基因
ifelse( deg_DESeq2$log2FoldChange > logFC_t,'up', #接上句else 否则:接下来开始判断那些P.Value<0.01的基因,再if 判断:如果logFC >1.5,则为up(上调)基因
ifelse( deg_DESeq2$log2FoldChange < -logFC_t,'down','stable') )#接上句else 否则:接下来开始判断那些logFC <1.5 的基因,再if 判断:如果logFC <1.5,则为down(下调)基因,否则为stable基因
)
deg_combat$g=ifelse(deg_combat$padj > pvalue_t,'stable', #if 判断:如果这一基因的P.Value>0.01,则为stable基因
ifelse( deg_combat$log2FoldChange > logFC_t,'up', #接上句else 否则:接下来开始判断那些P.Value<0.01的基因,再if 判断:如果logFC >1.5,则为up(上调)基因
ifelse( deg_combat$log2FoldChange < -logFC_t,'down','stable') )#接上句else 否则:接下来开始判断那些logFC <1.5 的基因,再if 判断:如果logFC <1.5,则为down(下调)基因,否则为stable基因
)
gplots::balloonplot(
table( deg_DESeq2[ids,'g'],
deg_combat[ids,'g'])
)
如下所示:
down stable up
down 100 33 0
stable 18 11860 42
up 0 14 35
两种不同的差异分析策略拿到的统计学显著的上下调基因列表重合度还不错,而且也可以进一步去看看它们的功能富集差异。
单细胞如何弄呢
常规转录组数据分析如果仅仅是为了拿到统计学显著的上下调基因列表,其实并不需要去除了批次效应后的表达量矩阵,因为后面的富集分析都是基于基因的。而且我们证明了两种不同的差异分析策略的一致性挺好的,所以无需担心。
但是如果是要热图可视化这些基因,就需要除了批次效应后的表达量矩阵啦。或者说是在你的热图里面标记清楚样品的分组以及批次等信息,也是可以一定程度的展现出来。
现在大家主要是做单细胞转录组数据分析,也会面临这样的”去批次效应”(batch effect removal)这个操作,而我们推荐的harmony确实是并不会修改原始的表达量矩阵的,那么就有一个问题,我们的差异分析就只能说针对原始的单细胞转录组表达量矩阵(几万个基因几万个细胞)吗?
其实单细胞转录组差异分析有一个问题,如果是不同单细胞亚群,其实完全无需理会这个批次效应啦,因为免疫细胞和上皮细胞这样的 差异是很容易展示出来的,但是如果是同一个单细胞亚群在不同处理的样品里面的差异就有点麻烦了,如果是简单的FindMarkers类似的函数,其实没办法抹去单细胞转录组个体差异 :
table(Idents(sce))
## 第一次差异分析在AA子集里面,看
sce_tmp = sce[,sce$g2=='AA']
table(sce_tmp$celltype,Idents(sce_tmp))
(tp = sort(unique(sce_tmp$celltype)) [-1])
degs_in_AA = lapply(tp, function(x){
tmp = FindMarkers(sce_tmp[,sce_tmp$celltype==x],ident.1 = 'SHR',
ident.2 = 'WKY')
tmp$batch = 'AA'
tmpr
})
names(degs_in_AA)=paste0('AA_',tp)
但是我们推荐的单细胞转录组差异分析其实是AggregateExpression后的矩阵,可以当做是一个传统的bulk转录组测序后的矩阵 :
sce.all = readRDS('./2-harmony/sce.all_int.rds')
sp='human'
load('./phe.Rdata')
identical(rownames(phe) , colnames(sce.all))
sce.all@meta.data = phe
sel.clust = "celltype"
sce.all <- SetIdent(sce.all, value = sel.clust)
table(sce.all@active.ident)
DimPlot(sce.all)
av <-AggregateExpression(sce.all ,
group.by = c("orig.ident","celltype"),
assays = "RNA")
av=as.data.frame(av[[1]])
head(av) # 可以看到是整数矩阵
#write.csv(av,file = 'AverageExpression-0.8.csv')
cg=names(tail(sort(apply(log(av+1), 1, sd)),1000))
pheatmap::pheatmap(cor(as.matrix(log(av[cg,]+1))))
save(av,file = 'av_for_pseudobulks.Rdata')