之前在在单细胞天地教程:表达矩阵逆转为10X的标准输出3个文件,详细介绍过 10X技术的单细胞转录组的3个标准文件,虽然说绝大部分文献提供其数据的时候并不是标准的文件名字,但是3个文件的文件名字还是通常会遵循以下模式:
- Feature / Gene-Barcodes Matrix 文件: 这个文件的命名通常包含了数据类型(例如基因表达量)和文件格式(例如稀疏矩阵)。一般情况下,这个文件名中可能包含 “matrix”、”gene_bc_matrix” 或类似的关键词。有时也会包含数据集的名称或样本编号。
- Barcode 文件: 这个文件通常命名为 “barcodes” 或者包含 “barcode” 关键词。里面有每个样品里面的每个细胞的标签信息,这个信息其实是无所谓的。
- Feature / Gene ID 文件: 这个文件通常命名为 “features”、”genes” 或包含 “gene” 关键词。取决于不同版本的cellranger定量结果。
需要把每个样品都整理成为3个标准文件,文件名字和文件格式如下所示:
所以很容易批量读取这样的文件,代码如下所示:
dir='GSE201048_RAW/outputs/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
tmp = Read10X(file.path(dir,pro ))
if(length(tmp)==2){
ct = tmp[[1]]
}else{ct = tmp}
sce =CreateSeuratObject(counts = ct ,
project = pro ,
min.cells = 5,
min.features = 300 )
return(sce)
})
也有一些文献给的是txt或者csv格式的,如下所示的文件:
10M 7 30 2019 GSM3984317_NO.1.expression_matrix.txt.gz
4.6M 7 30 2019 GSM3984318_NO.2.expression_matrix.txt.gz
5.3M 7 30 2019 GSM3984319_NO.3.expression_matrix.txt.gz
7.3M 7 30 2019 GSM3984322_NO.7.expression_matrix.txt.gz
13M 7 30 2019 GSM3984326_NO.12.expression_matrix.txt.gz
5.0M 7 30 2019 GSM3984330_NO.16.expression_matrix.txt.gz
14M 4 24 2020 GSM4495152_NO.20.expression_matrix.txt.gz
代码也是很简单的,批量读取,如下所示:
dir='GSE135045_RAW/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
f= file.path(dir,pro);print(f)
ct <- data.table::fread( f, data.table = F)
ct[1:4,1:4]
rownames(ct)=ct[,1]
ct=ct[,-1]
sce <- CreateSeuratObject(ct,project = gsub('.expression_matrix.txt.gz','',pro),
min.cells = 5,
min.features = 500) #后面就可以单细胞处理的标准流程啦
return(sce)
})
do.call(rbind,lapply(sceList, dim))
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples )
sce.all <- JoinLayers(sce.all)
但是我看到了一个比较狡猾的数据集(GSE133283),它官网给出来了的文件如下所示:
32M 6 24 2019 GSM3904816_Adult-1_gene_counts.tsv.gz
32M 6 24 2019 GSM3904817_Adult-2_gene_counts.tsv.gz
33M 6 24 2019 GSM3904818_Adult-3_gene_counts.tsv.gz
37M 6 24 2019 GSM3904819_Aged-1_gene_counts.tsv.gz
37M 6 24 2019 GSM3904820_Aged-2_gene_counts.tsv.gz
64M 6 24 2019 GSM3904821_EAE-1_gene_counts.tsv.gz
63M 6 24 2019 GSM3904822_EAE-2_gene_counts.tsv.gz
30M 6 24 2019 GSM3904823_Juvenile-1_gene_counts.tsv.gz
29M 6 24 2019 GSM3904824_Juvenile-2_gene_counts.tsv.gz
44M 6 24 2019 GSM3904825_Juvenile-3_gene_counts.tsv.gz
40M 6 24 2019 GSM3904826_Juvenile-4_gene_counts.tsv.gz
它看起来是每个样品一个独立的文本文件,但里面并不是行列式的表达量矩阵文件,读入简单肉眼看了看:
> f= file.path(dir,pro);print(f)
[1] "GSE133283_RAW/GSM3904816_Adult-1_gene_counts.tsv.gz"
ct <- data.table::fread( f, data.table = F)
> head(ct)
gene cell count
1 0610005C13Rik AACCGCGTCCGTTGCT 1
2 0610005C13Rik ATTATCCAGTTGTCGT 1
3 0610005C13Rik CAGTAACAGCAGCCTC 1
4 0610005C13Rik CATCGAAGTAGCGTAG 1
5 0610005C13Rik CCACTACTCACATGCA 1
6 0610005C13Rik CCTAAAGCAAGTAATG 1
> ct[1:4,1:4]
Error in `[.data.frame`(ct, 1:4, 1:4) : undefined columns selected
> dim(ct)
[1] 6182813 3
是稀疏矩阵的简化版,我略微思考了一下,做了一个简单的变幻:
library(reshape2)
tmp = dcast(ct,gene~cell)
tmp[1:4,1:4]
可以看到,很多NA,其实就是单细胞转录组里面的0值,需要替换一下:
> tmp[1:4,1:4]
gene AAACCTGAGATGTGTA AAACCTGAGGTACTCT AAACCTGAGTGTTAGA
1 0610005C13Rik NA NA NA
2 0610007N19Rik NA NA NA
3 0610007P14Rik 1 2 NA
4 0610009B14Rik NA NA NA
> dim(tmp)
[1] 21892 3664
这个时候我们可以借助于r编程语言里面的reshape2包的dcast函数进行数据转换,在 R 语言中,reshape2 包提供了 dcast() 函数,用于将数据框从长格式(long format)转换为宽格式(wide format)。长格式数据通常包含多行和少列,每行对应一个观察值,并且包含一个用于标识不同组的变量;而宽格式数据通常包含少行和多列,每行对应一个唯一的标识符,并且包含多个变量。最后的完整的代码是:
dir='GSE133283_RAW'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
f= file.path(dir,pro);print(f)
ct <- data.table::fread( f, data.table = F)
head(ct)
dim(ct)
#ct[1:4,1:4]
library(reshape2)
tmp = dcast(ct,gene~cell)
tmp[1:4,1:4]
ct = tmp
rownames(ct)=ct[,1]
ct=ct[,-1]
ct[is.na(ct)]=0
sce <- CreateSeuratObject(ct,project = gsub('_gene_counts.tsv.gz','',pro),
min.cells = 5,
min.features = 500) #后面就可以单细胞处理的标准流程啦
return(sce)
})
有了这个seurat的对象,后面就是我们常规的!代码在:(链接: https://pan.baidu.com/s/1pKEnPmWXi-pTab0WZUWzgg?pwd=a7s1) 相信大家很容易跟着去复现一次!
学徒作业
这个狡猾的数据集(GSE133283)对应的文章是:《Single-cell transcriptomics reveals functionally specialized vascular endothelium in brain》,文献里面的第一层次降维聚类分群如下所示:
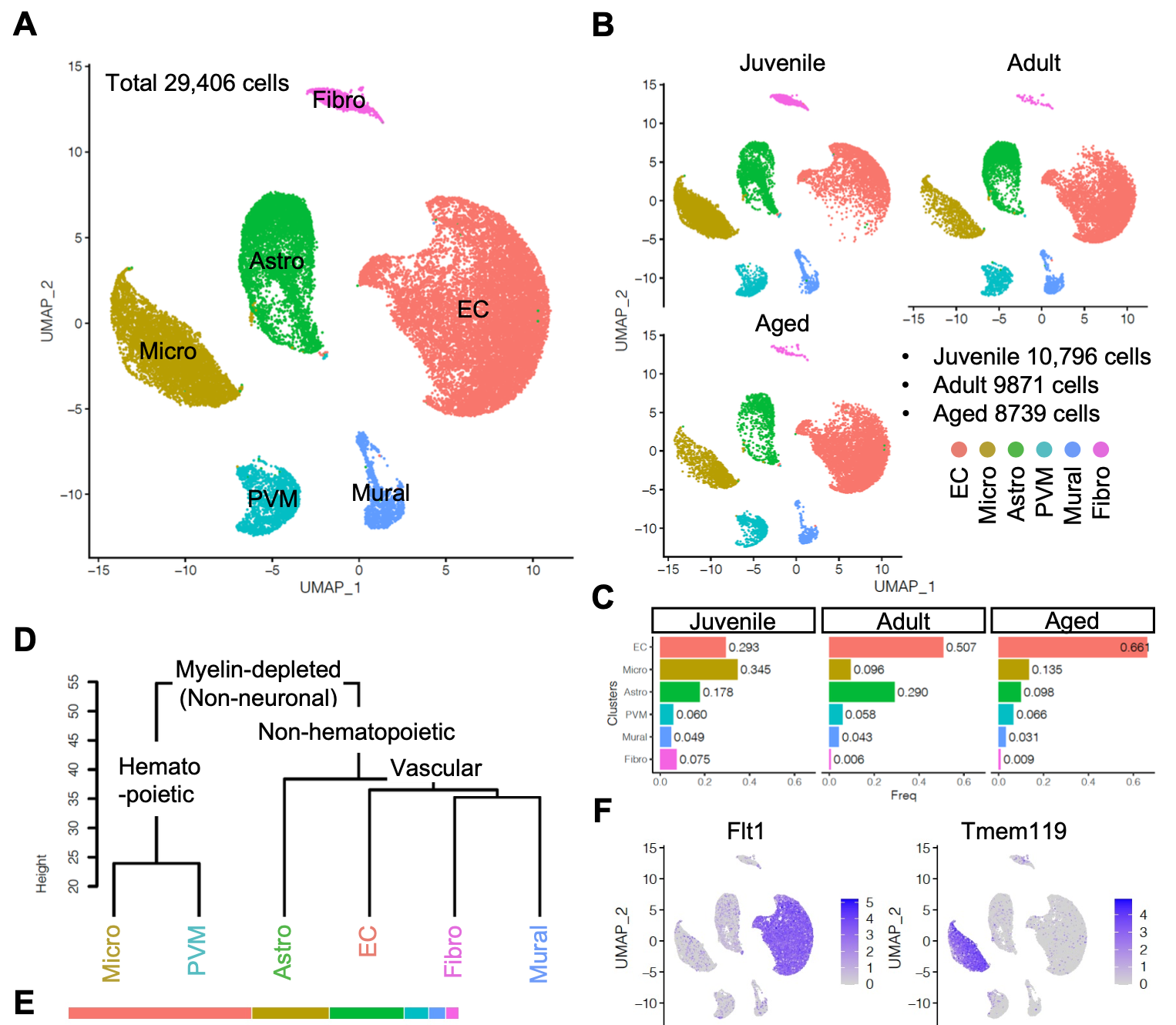
可以仔细看看文章里面的降维聚类分群参数,反正我使用标准代码跑了一下,没有文章那么清晰,不过我也解释过,这个肉眼可视化其实并没有那么重要,不影响分群后的各个亚群的生物学意义即可。
