本笔记会被收录于《生信技能树》公众号的《单细胞2024》专辑,而且我们从2024开始的教程都是基于Seurat的V5版本啦,之前已经演示了如何读取不同格式的单细胞转录组数据文件,如下所示:
- 初试Seurat的V5版本
- 使用Seurat的v5来读取多个10x的单细胞转录组矩阵
- 使用Seurat的v5来读取多个不是10x标准文件的单细胞项目
单细胞转录组早期有很多cns文章,但如今它已经是一个常规技术所以绝大部分实验设计就必然是“落入俗套”。那么这样的单细胞转录组项目也是有众所周知的常规数据分析策略啦,如果你不幸的继承了这样的“祖传”的单细胞转录组数据,也想简简单单发个文章而已,那么单细胞转录组的3种常规数据分析思路可能会对你有帮助啦。
我这里简单的分享一下自己看到的单细胞转录组的3种常规数据分析思路给大家。思路1:针对每个亚群的细分
早期的,比如2019年底的文章《Single-Cell RNA Sequencing to Dissect the Immunological Network of Autoimmune Myocarditis》,就是如此,它数据集是GSE142564,是小鼠心脏免疫细胞,数据分析基本上就第一层次降维聚类分群后的每个细分亚群同样的流程即可。而且因为是早期单细胞转录组数据分析,所以那个时候还不流行多样品的整合问题,只需要按照已知的标记基因把单细胞亚群合理的注释即可,比如下面的t细胞就散步在tsne二维图的各个地方:
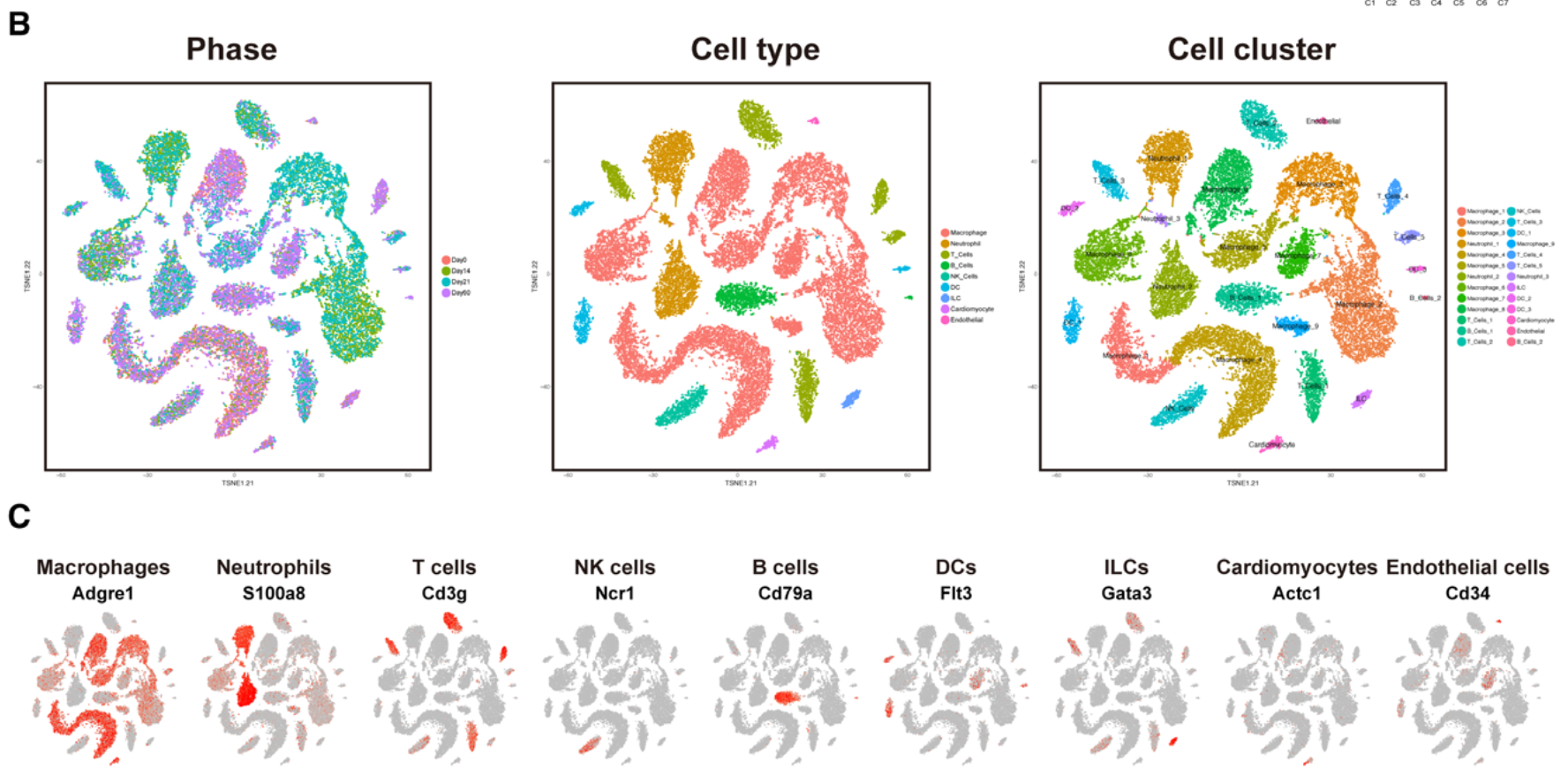
这些免疫细胞里面的髓系和t或者B的淋巴细胞细胞细分亚群可以细分,而文章也是确实是这样的做的: - B细胞细分亚群
- 髓系免疫细胞细分亚群
然后呢,最新的典范文献有 Hematol Oncol . 2023 Aug; 的:《Chemoresistance in acute myeloid leukemia: An alternative single‐cell RNA sequencing approach》 - 第一层次降维聚类分群后,比如:
- 然后每个亚群都是细分后看top基因以及其对应的功能,看分组(如果有)的细胞亚群比例变化以及差异分析全套
- 全局细胞通讯以及差异通讯事件
这个是没有对应的数据集公开获取,而且文章本身的分析“漏洞百出”,但是分组的实验设计本身是很“经典”的:13 untreated patients with de novo AML were enrolled and stratified into two groups: complete remission (CR; n = 8) and non-CR (n = 5).
再比如2021-GSE163558-胃癌淋巴结转移的文章,标题是:《Revealing the transcriptional heterogeneity of organ-specific metastasis in human gastric cancer using single-cell RNA Sequencing》,也是第一层次降维聚类分群后,针对每个亚群继续细分探索即可:
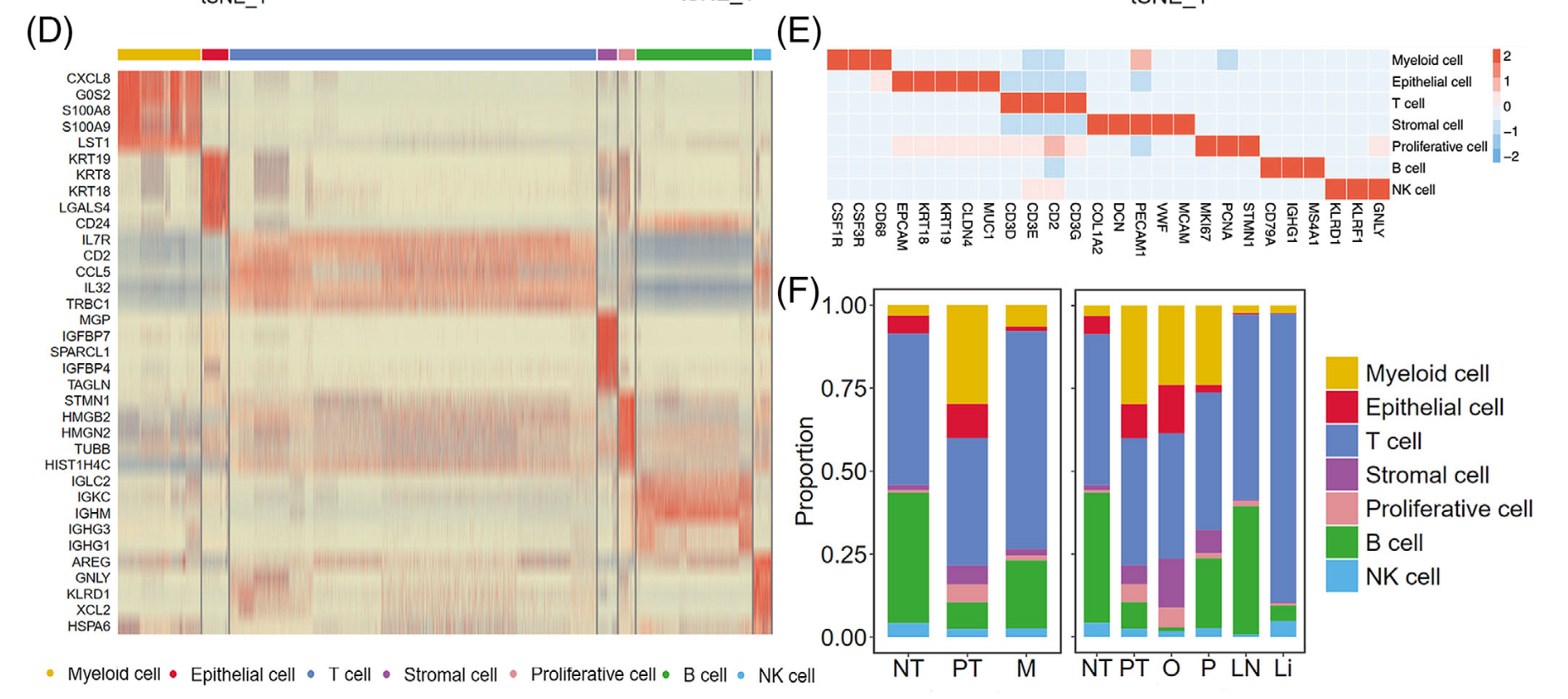
包括: - FIGURE 2 FoursubclustersofmalignantepithelialcellsinGCandtheircharacterisationassociatedwithmetastasis.(A)tSNEof1615 malignant and 128 non-malignant epithelial cells. (B) tSNE of the four malignant epithelial clusters G0–G3.
- FIGURE 3 VariousimmuneresponsesaremediatedbyTandBcellsduringGCprogression.
- FIGURE 4 EndothelialcellspromoteangiogenesisandcreateimmuneresistanceinGC.(
- FIGURE 5 iCAFscouldbeidentifiedintheTMEofGCandareassociatedwithtumourinvasion.
- FIGURE 6 MyeloidcellsareabundantduringGCprogression.
是不是超级简单啊,代码基本上都是复制粘贴即可哈,重点的单细胞亚群探索完毕就可以总体做表达量差异和细胞比例差异, T and B lymphocyte subclusters vary among different organ metastases.
后面可以做很多高级分析:
- 癌症单细胞的恶性肿瘤细胞判定新方法-SCEVAN
- 2款最适合单细胞数据分析的服务器配置
- 毛遂自荐成为你的单细胞顾问
- 10x官网下载pbmc3k数据集走RNA速率上下游分析实战
- pyscenic的转录因子分析结果展示之各个单细胞亚群特异性激活转录因子
- pyscenic的转录因子分析结果展示之5种可视化
思路2:针对一个特殊(目标)亚群细致探索
典范文献:《 Cux1+ proliferative basal cells promote epidermal hyperplasia in chronic dry skin disease identified by single-cell RNA transcriptomics 》
第一层次降维聚类分群后,比如:是8个簇,分别为成纤维细胞(FIB),角质形成细胞(KC),骨髓细胞(MYL), T细胞(TC),脂肪细胞(ADI),内皮细胞(ENDO),肥大细胞(MC)和血管平滑肌细胞(vSMC)
然后就是针对角质形成细胞(KC) 这个特殊(目标)亚群的细致分析:
- 继续细分成为5个亚群,包括棘细胞(SC)、基底细胞(BC)、皮脂腺细胞(SGC)、增殖型基底细胞(PBC)和颗粒细胞(GC)
- 每个亚群都是细分后看top基因以及其对应的功能,看分组(如果有)的细胞亚群比例变化以及差异分析全套
- 拟时序
- 转录因子
- 全局细胞通讯以及差异通讯事件
如下所示,也是标准的代码:
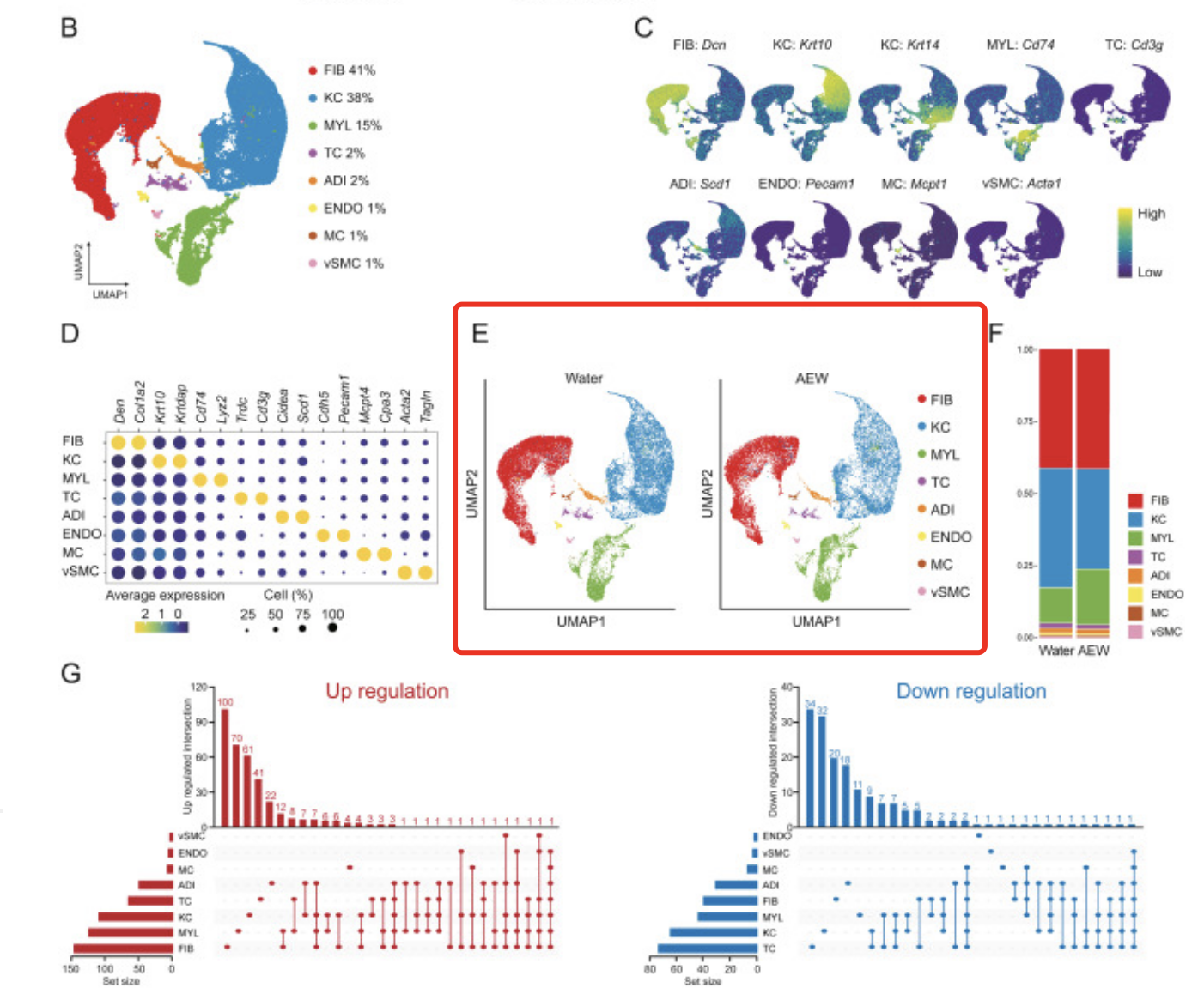
文章的配图就完完全全说明了他的分析情况: - Fig. 1. Cell type identification by single-cell RNA sequencing (scRNA-seq) analysis of dry skin in mice.
- Fig. 2. Transcriptional classification of mouse keratinocytes into five subpopulations.
- Fig. 3. Pseudotime trajectories analysis reveals keratinocyte differentiation dynamics in dry skin.
- Fig. 4. Novel CUT-like homeobox 1 (Cux1)-positive proliferative basal cells (PBC) subpopulation in dry skin.
- Fig. 5. Potential ligand-receptor interactions between proliferative basal cells with other cells.
- Fig. 6. Transcription factor CUT-like homeobox 1 (CUX1) promoted cell proliferation on keratinocytes. (
- Fig. 7. CUT-like homeobox 1 (Cux1)-positive proliferative basal cells were increased in psoriasis.
思路3:临床或者公共数据联合
其实这个思路的数据分析,都没必要自己测序了,有点浪费。因为目前海量的数据挖掘文章都是基于单细胞转录组公共数据去结合临床信息的:
- 联合同样的实验设计的bulk数据多分组差异分析,代表文献:
- 《Single-cell transcriptome profiling of the vaginal wall in women with severe anterior vaginal prolapse 》
- 《Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease 》
- 联合预后或者疗效等相关临床信息做模型,代表文献:《A TCF4-dependent gene regulatory network confers resistance to immunotherapy in melanoma》
- 联合分子分型,疾病进展等相关临床信息做模型,代表文献:《Epithelial cells activate fibroblasts to promote esophageal cancer development》