开始处理蛋白质组学数据之前,大家肯定是会好奇数据如何产生,它本质上也是具体的每个蛋白质在具体的每个样品的表达量检测而已。而且蛋白质组学技术肯定是不止质谱(Mass Spectrometry, MS),其它技术也会产生表达量矩阵,有了矩阵后的下游分析就大同小异了。所以我们推荐大家看两个综述来了解这个技术的前因后果哈。
基于质谱(MS)的蛋白质组学数据分析
这篇综述文章《Bioinformatics Methods for Mass Spectrometry-Based Proteomics Data Analysis》由Chen Chen等人撰写,发表在《International Journal of Molecular Sciences》上,主要讨论了基于质谱(MS)的蛋白质组学数据分析中的生物信息学方法。不过这个综述里面的蛋白质组学数据分析的上下游划分方式我不是很认可,我认为的:
- 上游应该是质谱产生raw数据然后经过搜库这个过程变成表达量矩阵以及矩阵的一些质量控制分析
- 下游就是基于表达量矩阵的各种差异分析富集分析基本上等同于转录组数据分析啦(表达量芯片或者转录组测序)
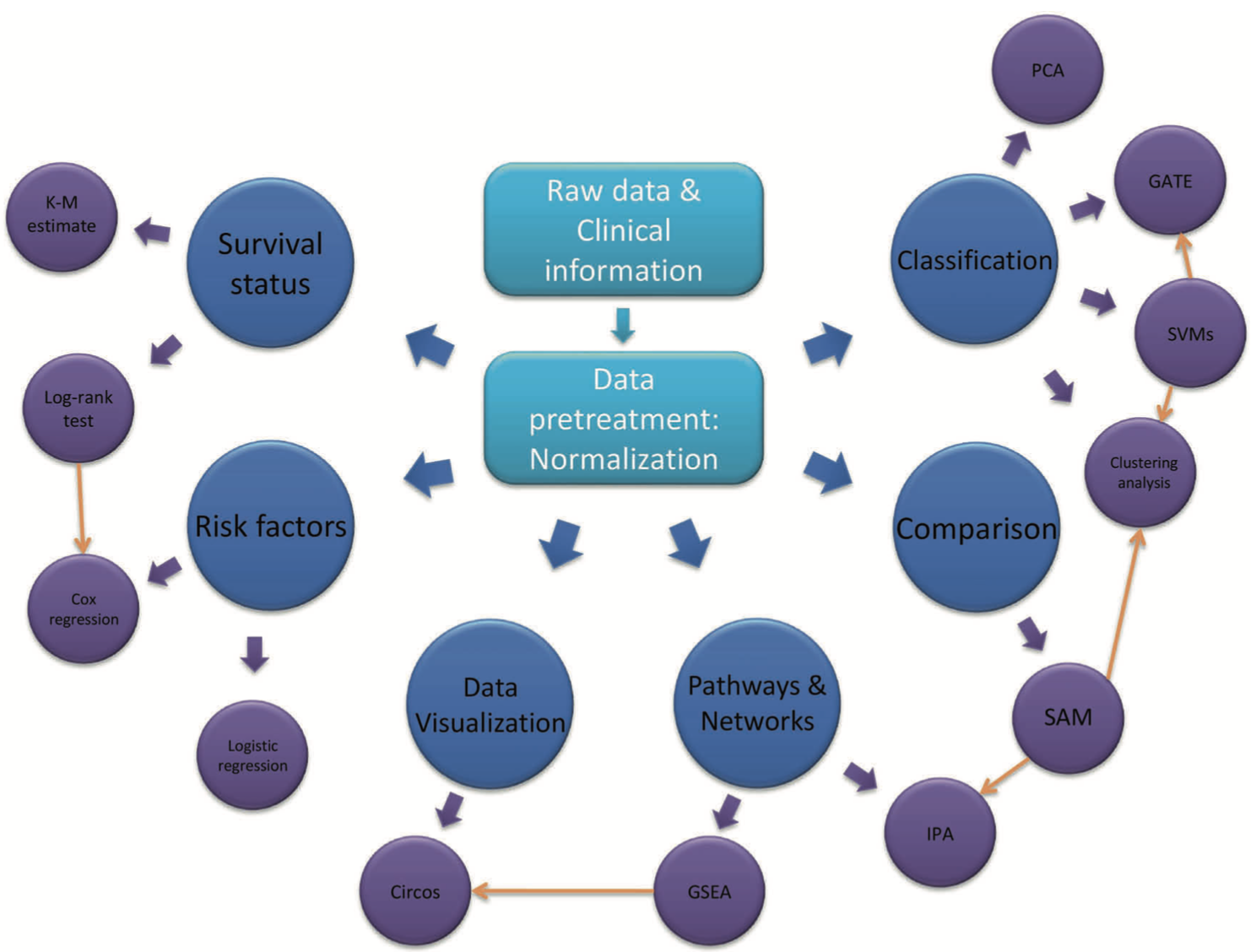
以下是这篇综述文章的核心内容概述:
- 蛋白质组学的重要性:蛋白质组学对于理解细胞机制、疾病进展以及基因型与表型之间的关系至关重要。
- 质谱技术的发展:质谱技术的进步使得研究人员能够对复杂生物系统中的蛋白质进行全面分析。
- 蛋白质鉴定与定量:介绍了用于处理原始质谱数据的工具和方法,包括肽段和蛋白质的鉴定及定量。
- 生物信息学工具:讨论了用于质谱基础蛋白质组学数据的生物信息学软件和平台,以及用于下游分析的不同统计和机器学习方法。
- 数据分析方法:文章重点介绍了数据预处理、统计分析和富集分析中使用的算法和工具,并讨论了机器学习在蛋白质组学研究中的应用。
- 蛋白质相互作用网络:探讨了如何使用定量蛋白质数据来重建蛋白质相互作用和信号网络。
- 实验策略:讨论了基于质谱的蛋白质组学的典型实验策略,包括自下而上的(bottom-up)和自上而下的(top-down)方法。
- 软件工具:提供了用于肽段和蛋白质鉴定的软件工具列表,包括数据库搜索算法、去新肽序列分析工具、混合鉴定方法和其他与蛋白质/PTM鉴定相关的软件。
- 数据标准化和缺失值处理:讨论了数据预处理和标准化的重要性,包括缺失值的处理方法。
- 统计分析:介绍了用于定量蛋白质数据的统计分析方法,包括t-test、ANOVA和线性模型。
- 富集分析:讨论了如何使用富集分析来识别在预定义基因集中过度表达的基因或蛋白质。
- 机器学习方法:探讨了机器学习在蛋白质组学数据分析中的应用,包括监督学习和无监督学习。
- 蛋白质网络重建:讨论了系统生物学或网络生物学中蛋白质-蛋白质相互作用(PPI)网络和信号网络的重要性。
- 未来展望:文章最后讨论了MS基础蛋白质组学的未来发展方向,包括技术进步、多组学数据整合和生物信息学工具的更新。
值得注意的是上游数据分析里面的蛋白质鉴定与定量是两个不同的过程,但是在实践中,蛋白质鉴定和定量通常是联合进行的。例如,在质谱实验中,首先通过MS/MS数据进行蛋白质鉴定,然后利用相应的肽段信号强度或特定的定量标签来量化蛋白质的表达水平。这种结合使用鉴定和定量的方法可以提供关于蛋白质在不同条件下表达模式的详细信息,从而有助于理解生物学过程和疾病机制。 - 蛋白质鉴定(Protein Identification):
- 目的是确定样品中存在的蛋白质的身份。这通常涉及将质谱(MS)产生的肽段的质量和序列信息与数据库中的已知蛋白质序列进行比对。
- 鉴定过程依赖于质谱数据中的肽段指纹图谱或肽段序列的串联质谱(MS/MS)数据,通过数据库搜索来匹配候选蛋白质。
- 蛋白质鉴定可以揭示样品中存在的特定蛋白质或蛋白质的特定形式(如翻译后修饰的蛋白质)。
- 蛋白质定量(Protein Quantitation):
- 目的是测量样品中蛋白质的相对或绝对丰度。定量可以是定性的(比较不同样品中蛋白质的相对表达水平)或绝对定量的(测量蛋白质的拷贝数或浓度)。
- 定量方法包括使用稳定同位素标记的标签(如iTRAQ、TMT、SILAC)或无标记方法(如基于肽段或蛋白质离子强度的LFQ或基于标签的定量)。
- 定量数据可以用于比较不同生物学状态下的蛋白质表达变化,如健康与疾病状态,或响应不同实验处理的条件。
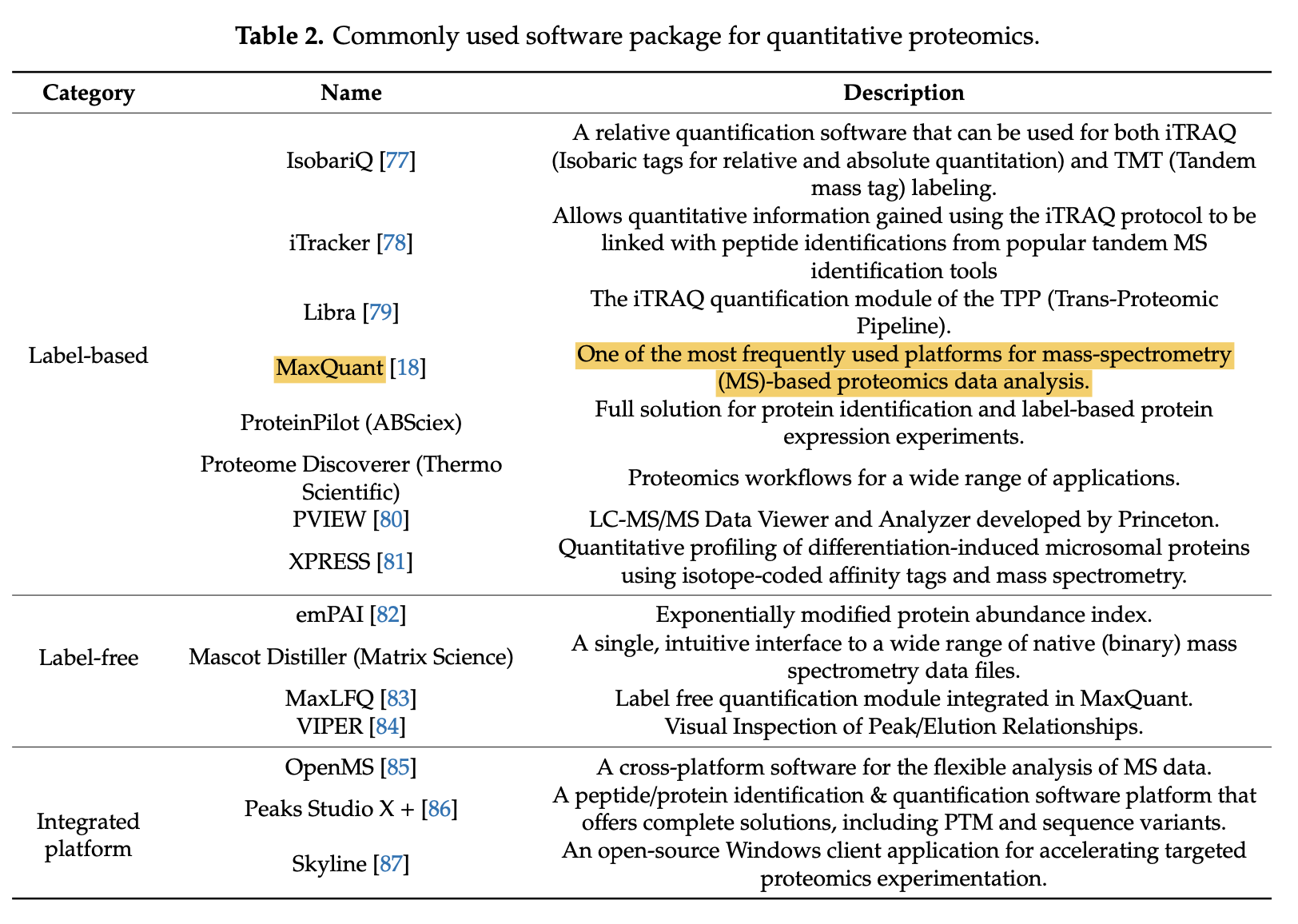
对大家来说,耳熟能详的软件就是 MaxQuant 啦, 如下所示:
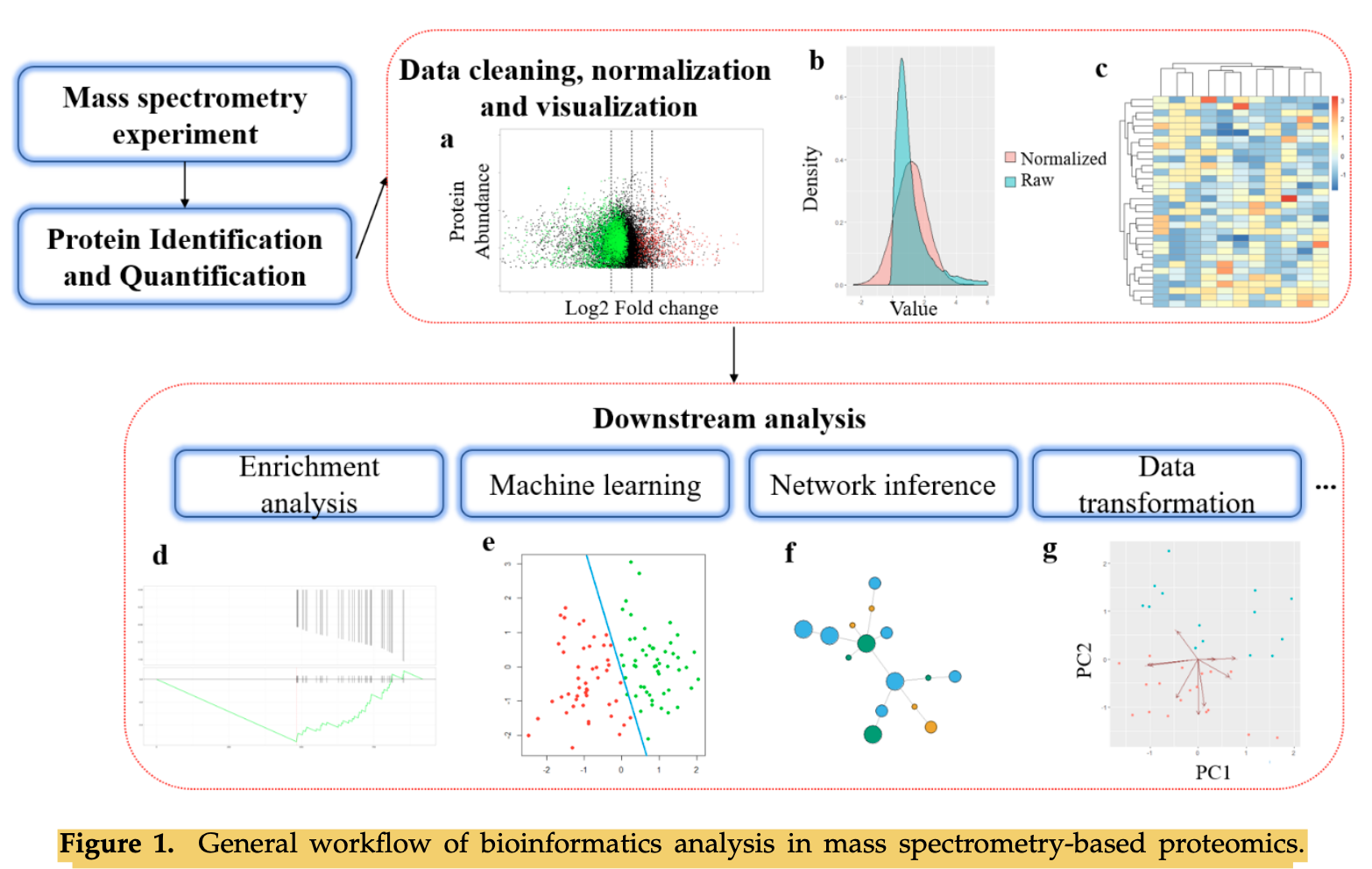
然后,前面我们提到过下游就是基于表达量矩阵的各种差异分析富集分析基本上等同于转录组数据分析啦(表达量芯片或者转录组测序),但是基于质谱(MS)的蛋白质组学数据有一个特点是有缺失值,往往是需要插补的。
一个蛋白质组学数据分析上下游的衔接步骤可以是MaxQuant拿到矩阵然后走DEP这个r包,如下所示:
- The raw mass spectrometry data were first analyzed using MaxQuant (Cox and Mann, Nat Biotech 2007) and the resulting “proteinGroups.txt” file is used as input for the downstream analysis.
- It requires tabular input (e.g. txt files) as generated by quantitative analysis softwares of raw mass spectrometry data, such as MaxQuant or IsobarQuant.
- Functions are provided for data preparation, filtering, variance normalization and imputation of missing values, as well as statistical testing of differentially enriched / expressed proteins.
高通量蛋白质组学技术
这篇综述文章《High-throughput proteomics: a methodological mini-review》由Miao Cui、Chao Cheng和Lanjing Zhang撰写,发表在《Laboratory Investigation》上,主要讨论了高通量蛋白质组学技术在生物医学研究中的应用,并概述了现有的和新兴的高通量蛋白质组学方法,包括:
- 高通量蛋白质组学技术:包括质谱(Mass Spectrometry, MS)、蛋白质途径阵列(Protein Pathway Array, PPA)、下一代组织微阵列(Next-generation Tissue Microarrays, ngTMAs)、单细胞蛋白质组学(Single-Cell Proteomics, SCP)、单分子蛋白质组学(Single-Molecule Proteomics, SMP)、Luminex、Simoa和Olink蛋白质组学等。
- 计算方法和统计算法:讨论了重要的计算方法和统计算法,这些方法可以最大化蛋白质组数据与临床和其他“组学”数据的挖掘。
- 技术发展:总结了过去十年中蛋白质组学方法的发展,以及它们在疾病诊断、精准医学和药物发现中的潜在应用。
- 蛋白质组学的重要性:强调了蛋白质组学在后基因组时代生物医学研究中的关键作用,尤其是在理解疾病分子机制和发现新的生物标志物方面。
- 蛋白质组学数据的解释:提到了人类蛋白质组项目(Human Proteome Project)和蛋白质组学数据解释的指导原则。
- 蛋白质组学技术的临床应用:讨论了如何将蛋白质组学技术从基础研究转化为临床应用,以及这一过程中的挑战和限制。
- 统计和算法:介绍了用于处理大量数据的新型高通量方法和基于机器学习的算法,包括监督学习和无监督学习方法。
- 数据预处理:强调了数据预处理中标准化的重要性,以及如何处理大数据以提高数值稳定性和模型拟合。
- 单细胞蛋白质组学:作为一门新兴技术,单细胞蛋白质组学专注于单细胞水平上的蛋白质分析,有望与单细胞转录组学互补,共同推进对单细胞生物学的理解。
- 未来展望:尽管蛋白质组学在发展过程中面临挑战,但它正朝着更深入的单细胞生物学和个体化精准医学方向发展,这将推动基础研究和临床实践达到新的高度。
文章强调了高通量蛋白质组学技术的进步不仅有助于更好地理解病理学的分子机制,而且还有助于识别特定疾病的特征信号网络。此外,文章还讨论了将蛋白质组学应用于疾病诊断的进展、当前挑战和未来展望。
下游分析五花八门,但是综述里面汇总的每个步骤都不是蛋白质组特有的哦: