在生信技能树公众号看到了练习题在:9个小鼠分成3组后取36个样品做转录组测序可以做多少组合的差异分析,需要读取这个表达量矩阵完成里面的层次聚类和组合pca分析。上游的定量过程是需要服务器的,这里省略,我们主要是演示一下多分组表达量矩阵的层次聚类和组合pca分析。
读取表达量矩阵以及样品分组信息
读取上游定量结果
# 魔幻操作,一键清空
rm(list = ls())
options(stringsAsFactors = F)
library(data.table)
library(stringr)
rawcount <- read.table("counts/all.id.txt",row.names = 1,
sep = "\t", header = T)
head(colnames(rawcount))
colnames(rawcount)=str_split(colnames(rawcount),'[.]',simplify = T)[,6]
#rawcount <-rawcount[,c(-3,-9)]
mat <- rawcount[,6:ncol(rawcount)]
mat[1:4,1:4]
keep_feature <- rowSums (mat > 1) > 1
ensembl_matrix <- mat[keep_feature, ]
上面的结果通常是ensembl数据库的id,需要转换为人类可以看得懂的symbol名字。
library(AnnoProbe)
head(rownames(ensembl_matrix))
ids=annoGene(rownames(ensembl_matrix),'ENSEMBL','mouse')
head(ids)
ids=ids[!duplicated(ids$SYMBOL),]
ids=ids[!duplicated(ids$ENSEMBL),]
symbol_matrix= ensembl_matrix[match(ids$ENSEMBL,rownames(ensembl_matrix)),]
rownames(symbol_matrix) = ids$SYMBOL
#symbol_matrix= ensembl_matrix
symbol_matrix[1:4,1:4]
可以看到矩阵修改前后主要是基因名字的区别:
> symbol_matrix[1:4,1:4]
SRR6789051 SRR6789052 SRR6789053 SRR6789054
Xkr4 608 617 991 1323
Gm18956 10 5 8 12
Gm37180 89 14 65 79
Gm37363 41 1 26 64
> mat[1:4,1:4]
SRR6789051 SRR6789052 SRR6789053 SRR6789054
ENSMUSG00000102628 25 23 11 5
ENSMUSG00000100595 2 0 0 0
ENSMUSG00000097426 1 2 0 0
ENSMUSG00000104478 0 0 0 0
关于Ensembl ID和Gene Symbol的区别:
- Ensembl ID:是由Ensembl数据库分配给每个基因和转录本的唯一标识符。它们通常用于自动化的生物信息学分析和数据库查询,因为它们提供了一种稳定且唯一的方法来引用特定的基因或转录本2。
- Gene Symbol:是基因的缩写形式,通常由大写字母组成,是基因的人类可读形式。Gene Symbol由各种命名委员会提供,如HGNC(人类基因命名委员会)负责人类基因的命名。这些符号更易于人类记忆和引用,在科学文献和交流中最常用。
需要注意的是,有时一个Gene Symbol可能对应多个Ensembl ID,因为一个基因可能有多个体细胞或同源基因。此外,由于基因命名的变化,一个基因可能有多个别名,因此在使用Gene Symbol时要特别小心。
在进行基因表达量矩阵的分析时,将Ensembl ID转换为Gene Symbol可以使结果更易于解释和共享,因为研究人员通常更熟悉Gene Symbol而不是数据库特定的ID。
有了表达量矩阵还不够,还需要分组信息,可以看到上面的样品名字目前也是id: SRR6789051 SRR6789052 SRR6789053 SRR6789054 也是需要加上合理的注释信息。
group <- read.table("./SraRunTable.txt",
header = T,sep = ",")
colnames(group)
# 提取表达矩阵对应的样本表型信息
group <- group[match(colnames(symbol_matrix),group$Run),
c("Run","tissue","pain_model")]
head(group)
table(group[,2:3])
# group_list=ifelse(grepl('Healthy',group$tissue),'control','case' )
# table(group_list)
# group_list
# group_list = factor(group_list,levels = c('control','case' ))
group_list=paste(group[,2 ],group[,3 ],sep = ' ')
table(group_list)
save(symbol_matrix,group_list,file = 'symbol_matrix.Rdata')
可以看到 3X4X3=36样品,详见::9个小鼠分成3组后取36个样品做转录组测序可以做多少组合的差异分析 ;
> head(group)
Run tissue pain_model
1 SRR6789051 brain CTR
2 SRR6789052 brain CTR
3 SRR6789053 brain CTR
4 SRR6789054 brain CFA
5 SRR6789055 brain CFA
6 SRR6789056 brain CFA
> table(group[,2:3])
pain_model
tissue CFA CTR SNI
blood 3 3 3
brain 3 3 3
dorsal root ganglia 3 3 3
spinal cord 3 3 3
有了上面的表达量矩阵,以及对应的样品分组信息,就可以做下面的简单的质量控制了,通常是层次聚类和主成分分析。
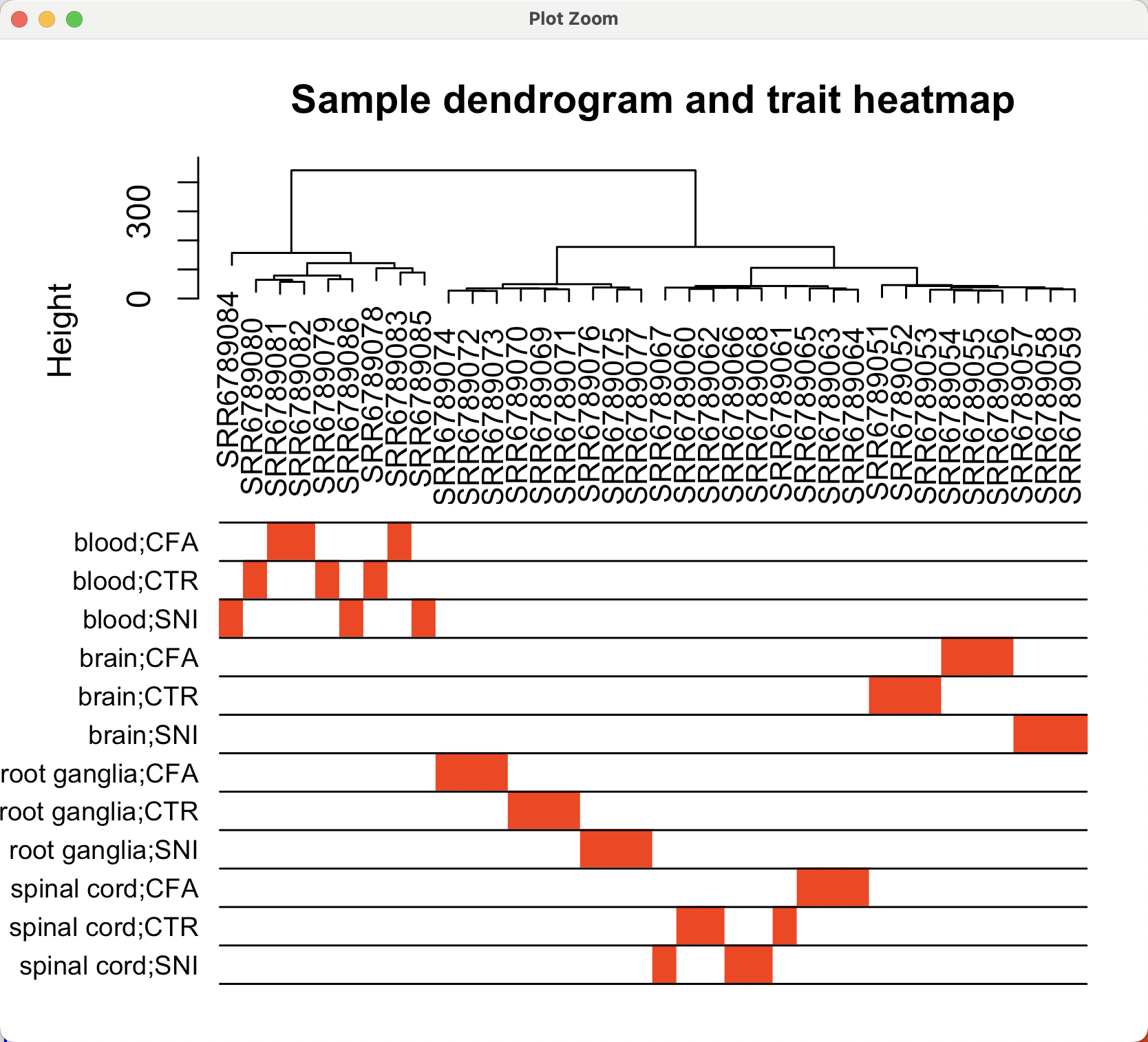
做层次聚类
表达量矩阵的层次聚类是一种用于分析和可视化基因表达数据的统计方法。在生物信息学和基因表达分析中,层次聚类可以帮助研究者根据基因表达模式将基因或样本分组,从而揭示不同样本间的相似性和差异性。
如何理解层次聚类:
- 相似性度量:层次聚类首先需要定义一个相似性度量来评估基因或样本之间的接近程度。对于基因,这通常是它们表达量的相关性;对于样本,这可能是样本间基因表达的整体相似度。
- 聚合过程:通过递归地合并最相似的基因或样本对,层次聚类构建了一个聚类树,也称为“树状图”或“谱系图”。每次迭代中,最相似的一对聚类被合并成一个新的聚类,然后这个新聚类再与其它聚类比较相似性。
- 距离计算:在每次合并后,需要计算新聚类与其他聚类之间的距离。常用的距离计算方法包括欧氏距离、曼哈顿距离、皮尔逊相关系数等。
- 剪枝形成聚类:通过设定一个距离阈值,可以决定在树状图的哪一点“剪枝”,即停止合并过程,从而形成最终的聚类。这个阈值可以是固定的,也可以是动态计算的。
- 结果解释:层次聚类的结果通常以树状图的形式展示,树状图的每个分支代表一个聚类,而分支的连接点则表示聚类合并的步骤。在基因表达分析中,树状图可以帮助识别具有相似表达模式的基因群,这些基因群可能涉及相同的生物学过程或功能。
- 生物学意义:层次聚类揭示的基因或样本的聚类模式,可以为进一步的实验设计、功能注释和生物标志物的发现提供线索。
library(WGCNA)
datExpr=t(log2(edgeR::cpm(symbol_matrix)+1))
sampleTree2 = hclust(dist(datExpr), method = "average")
# 用颜色表示表型在各个样本的表现: 白色表示低,红色为高,灰色为缺失
group_list=factor(group_list)
datTraits=as.data.frame(model.matrix(~0+ group_list))
rownames(datTraits)=rownames(datExpr)
colnames(datTraits)=levels(group_list)
traitColors = numbers2colors(datTraits, signed = FALSE)
# 把样本聚类和表型绘制在一起
plotDendroAndColors(sampleTree2, traitColors,
groupLabels = names(datTraits),
main = "Sample dendrogram and trait heatmap")
如下所示:
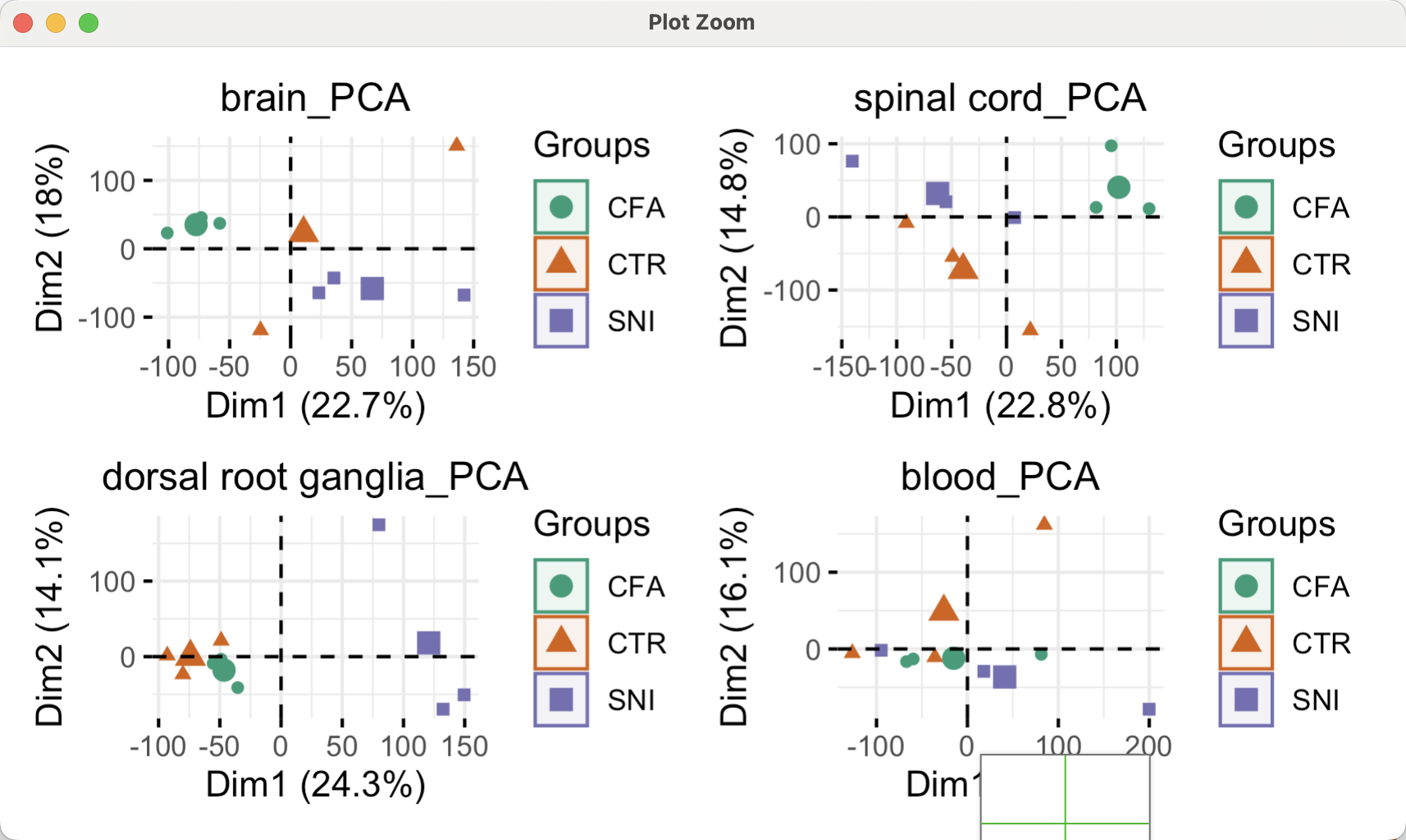
做pca分析
如果是真的整个表达量矩阵进行pca分析,那么就有点麻烦了,一张图上面很难完全展示清楚不同分组的关系,所以文章才会选择做四次pca分析啦,全部的代码如下所示:
# 魔幻操作,一键清空
rm(list = ls())
options(stringsAsFactors = F)
library(ggplot2)
library(ggstatsplot)
library(ggsci)
library(RColorBrewer)
library(patchwork)
library(ggplotify)
load(file = 'symbol_matrix.Rdata')
# https://mp.weixin.qq.com/s/bGclqc3qbpvm6rvwQtDWTg
colnames(symbol_matrix)
symbol_matrix[1:4,1:4] ## 基因名字的样品,矩阵
dat = log2(edgeR::cpm(symbol_matrix)+1)
dat[1:4,1:4]
boxplot(dat,las=2)
table(group_list)
library(stringr)
phe=as.data.frame(str_split(group_list,';',simplify = T))
table(phe)
my_draw_pca <- function(my_dat,my_pro,my_group_list){
# my_pro='all'
exp=t(my_dat)#画PCA图时要求是行名时样本名,列名时探针名,因此此时需要转换
exp=as.data.frame(exp)#将matrix转换为data.frame
dat.pca <- PCA(exp , graph = FALSE)
this_title <- paste0(my_pro,'_PCA')
p2 <- fviz_pca_ind(dat.pca,
geom.ind = "point", #c( "point", "text" ), # show points only (nbut not "text")
col.ind = my_group_list, # color by groups
palette = "Dark2",
addEllipses = TRUE, # Concentration ellipses
legend.title = "Groups")+
ggtitle(this_title)+
theme(plot.title = element_text(size=12,hjust = 0.5))
p2
}
tissues = unique(phe[,1])
p_list <- lapply(tissues, function(x){
#x = tissues[1]
print(x)
kp = phe[,1]==x;table(kp)
my_dat=dat[,kp]
my_pro=x
my_group_list=phe[kp,2]
my_draw_pca(my_dat,my_pro,my_group_list)
})
p_list[[1]]
p_list[[2]]
p_list[[3]]
p_list[[4]]
library(patchwork)
wrap_plots(p_list)
4次主成分分析结果组合起来如下所示: