学员在交流群反馈在处理一个2020的文章里面的GSE155513和GSE155512这两个单细胞转录组数据集遇到了两个物种数据集整合失败问题。
这两个数据集分别是人和鼠的SMC异质性探索的,文献标题是:《Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human》,可以看到GSE155513和GSE155512这两个单细胞转录组表达量矩阵是可以很好的整合:
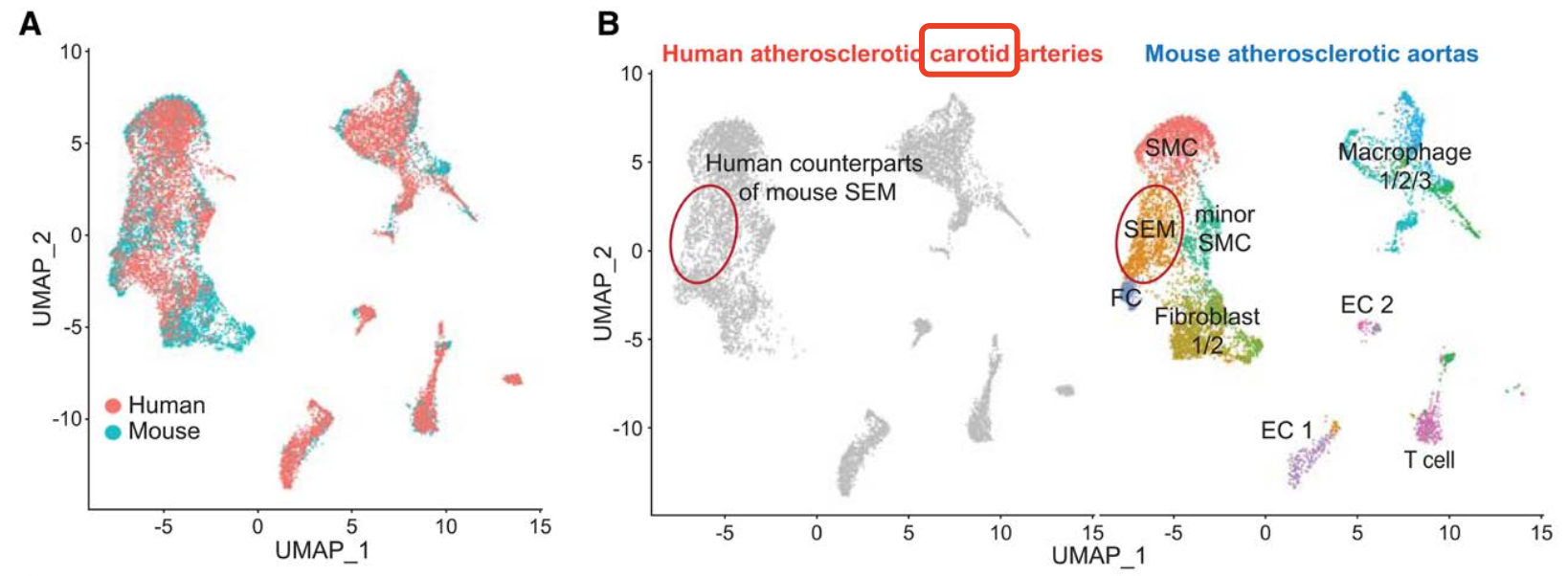
其中小鼠的样品比较多:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE155513
GSM4705592_RPS003_matrix.txt.gz 6.9 Mb
GSM4705593_RPS004_matrix.txt.gz 6.0 Mb
GSM4705594_RPS011_matrix.txt.gz 5.1 Mb
GSM4705595_RPS012_matrix.txt.gz 2.7 Mb
GSM4705596_RPS007_matrix.txt.gz 9.8 Mb
GSM4705597_RPS008_matrix.txt.gz 5.0 Mb
GSM4705598_RPS001_matrix.txt.gz 11.4 Mb
GSM4705599_RPS002_matrix.txt.gz 5.0 Mb
GSM4705600_RPS017_matrix.txt.gz 6.9 Mb
GSM4705601_RPS018_matrix.txt.gz 4.0 Mb
GSM4705602_RPS013_matrix.txt.gz 7.6 Mb
GSM4705603_RPS014_matrix.txt.gz 3.3 Mb
GSM4705604_RPS015_matrix.txt.gz 8.2 Mb
GSM4705605_RPS016_matrix.txt.gz 4.4 Mb
然后人类的是3个样品:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE155512
GSM4705589_RPE004_matrix.txt.gz 6.8 Mb
GSM4705590_RPE005_matrix.txt.gz 9.1 Mb
GSM4705591_RPE006_matrix.txt.gz 6.1 Mb
两个单细胞转录组表达量矩阵都是可以独立的降维聚类分群哦,然后有各自的的Seurat对象,接下来就试试看整合,代码如下所示:
mouse = readRDS('../GSE155513-鼠/2-harmony/sce.all_int.rds')
mouse$study = 'mouse'
table(mouse$orig.ident)
human = readRDS('../GSE155512-人/2-harmony/sce.all_int.rds')
human$study = 'human'
table(human$orig.ident)
mouse_ct = mouse@assays$RNA$counts
rownames(mouse_ct)=toupper(rownames(mouse_ct))
human_ct = human@assays$RNA$counts
ids=intersect(rownames(human_ct),rownames(mouse_ct))
mouse_ct = mouse_ct[ids,]
human_ct = human_ct[ids,]
可以看到的是我简简单单的把小鼠表达量矩阵的基因名字修改为了大写的,因为小鼠基因的命名规则通常包括将所有字母转换为小写,这与人类基因的命名规则不同,后者通常以大写字母开头。其实在进行跨物种的基因研究时,研究人员需要仔细核对基因的命名和序列信息,以确保研究的准确性。可以使用如Ensembl、UniProt或NCBI Gene等数据库来获取不同物种中基因的准确信息。
简简单单的把小鼠表达量矩阵的基因名字修改为了大写肯定是有很多基因会损失掉,比如人类:TP53(肿瘤蛋白p53)和小鼠:Trp53(与人类TP53同源)就基因名字不一样了,而不仅仅是大小写问题哦。
所以我对两个表达量矩阵取了共有基因的交集,然后就可以合并这两个矩阵啦, 如下所示:
sceList = list(
mouse = CreateSeuratObject(
counts = mouse_ct,
meta.data = mouse@meta.data
),
human = CreateSeuratObject(
counts = human_ct ,
meta.data = human@meta.data
)
)
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = c('mouse','human') )
names(sce.all@assays$RNA@layers)
sce.all[["RNA"]]$counts
# Alternate accessor function with the same result
LayerData(sce.all, assay = "RNA", layer = "counts")
sce.all <- JoinLayers(sce.all)
dim(sce.all[["RNA"]]$counts )
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all$orig.ident)
接下来就是常规的降维聚类分群哦,但是我们默认的流程里面的针对的仅仅是样品层面的整合,代码如下所示;
seuratObj <- RunHarmony(input_sce, c("orig.ident","study"))
其实会看到如下所示的警告信息:
Harmony 1/10
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
Harmony 2/10
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
Harmony 3/10
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
Harmony converged after 3 iterations
Warning: The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric
而且在后面的降维聚类分群也可以看到其实是整合效果不好, 两个物种仍然是泾渭分明的, 如下所示:
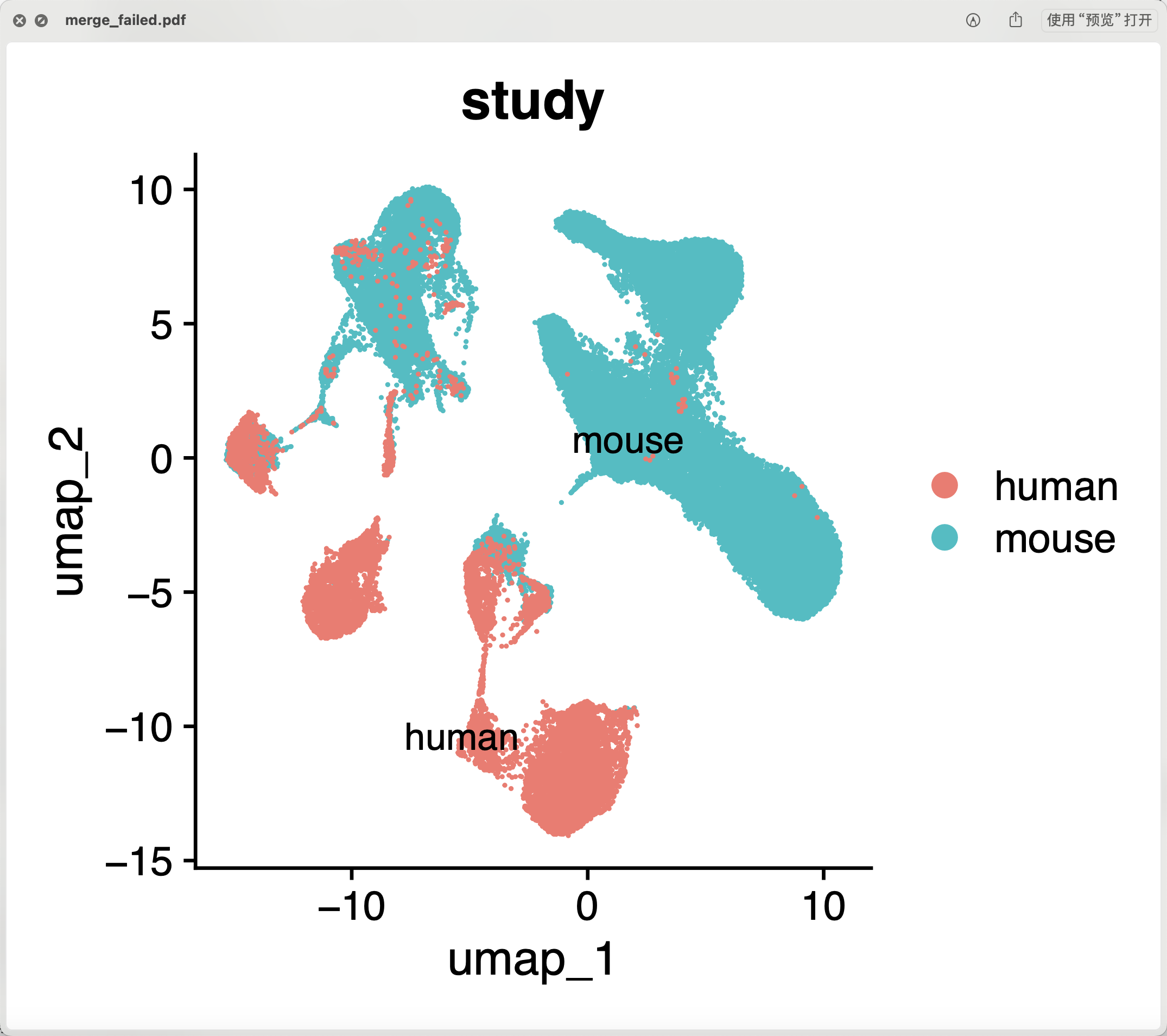
但是一般人都会忽略它,其实是RunHarmony函数可以修改参数的,比如同时抹去样品和数据集的差异,代码如下所示;
seuratObj <- RunHarmony(input_sce, c("orig.ident","study"))
可以看到这时候harmony走了10轮 :
**************************************************|
Harmony 10/10
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
这个时候,两个物种就比较好的整合在一起啦:
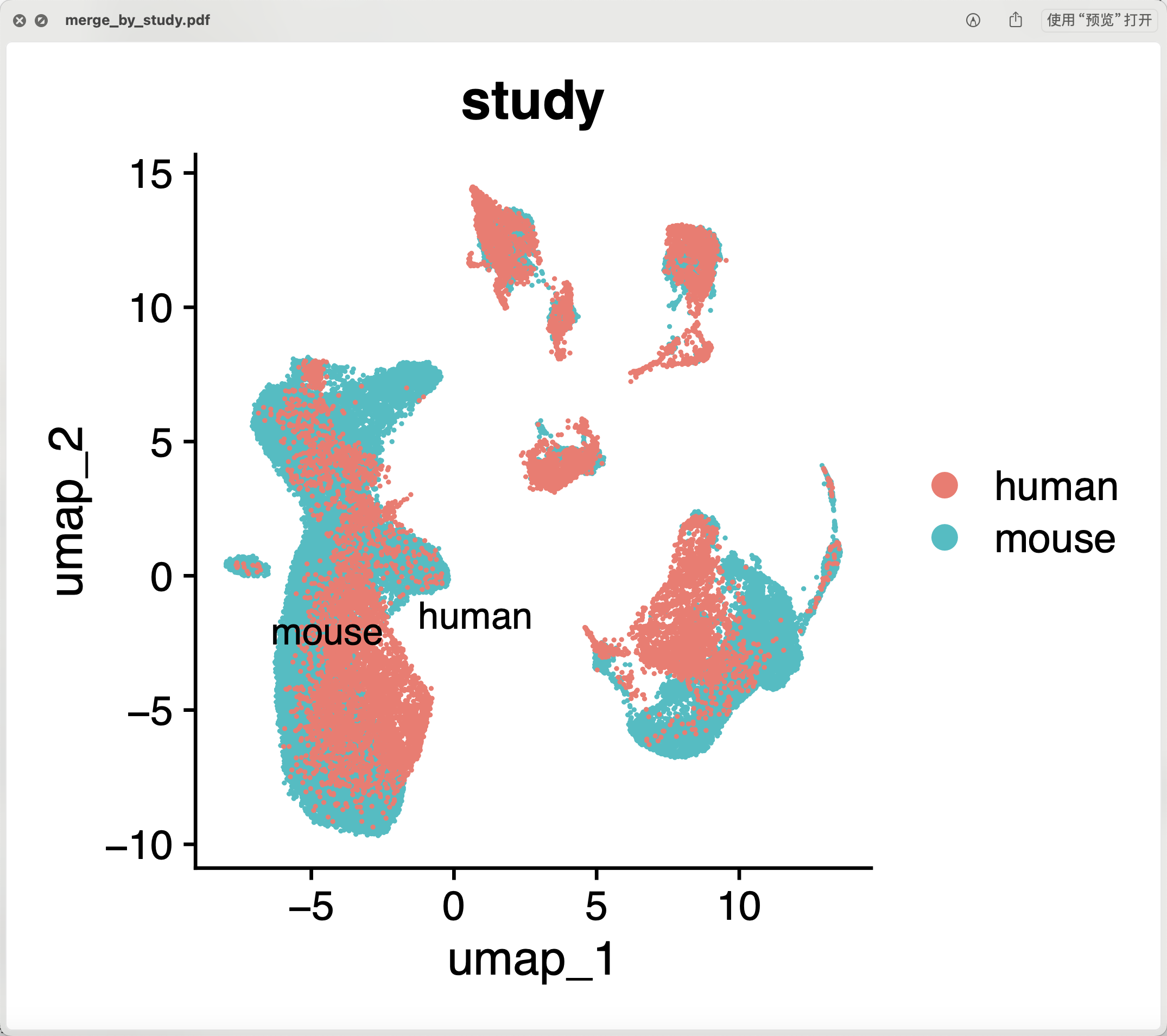
而且也是可以比较好的进行亚群的命名,跟原文一样的有两个泾渭分明的内皮细胞,然后就是t细胞和巨噬细胞代表的淋巴细胞和髓系免疫细胞啦 ,同样的文献里面的巨噬细胞和平滑肌细胞的界限也是模糊不清:
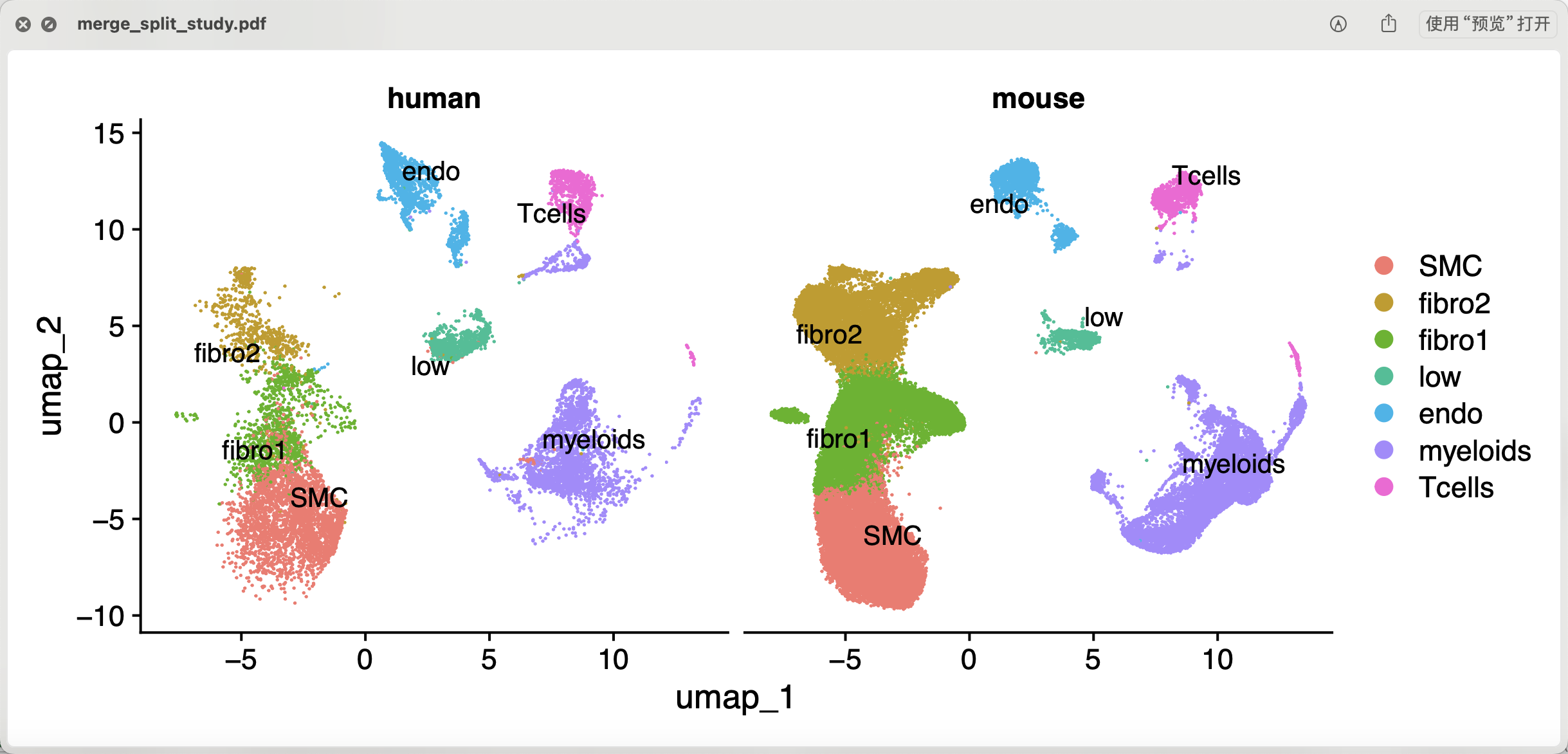
全部的代码,我整理好了放在百度云网盘里面,付费获取哈:( —来自百度网盘超级会员v9的分享 )
链接: https://pan.baidu.com/s/1wAkpTrNP-OfPVQfEb5q5_w?pwd=6asf 提取码: 6asf