前面我们推荐了方法学:使用singleR基于自建数据库来自动化注释单细胞转录组亚群,广受好评,然后马上就有小伙伴留言说这个功能跟Seurat的TransferData函数类似,我就马不停蹄的尝试了一下:
同样的数据集
这个GSE206528的单细胞转录组数据集,很容易构建成为Seurat对象。仍然是走常规的单细胞转录组降维聚类分群代码,可以看 链接: https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh ,基本上大家只需要读入表达量矩阵文件到r里面就可以使用Seurat包做全部的流程。批量读取它的9个文件的代码如下所示:
dir='GSE206528_RAW/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
ct=fread( file.path(dir,pro ) ,data.table = F)
ct[1:4,1:4]
rownames(ct)=ct[,1]
ct=ct[,-1]
sce =CreateSeuratObject(counts = ct ,
project = pro ,
min.cells = 5,
min.features = 300 )
return(sce)
})
do.call(rbind,lapply(sceList, dim))
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples )
names(sce.all@assays$RNA@layers)
sce.all[["RNA"]]$counts
# Alternate accessor function with the same result
LayerData(sce.all, assay = "RNA", layer = "counts")
sce.all <- JoinLayers(sce.all)
dim(sce.all[["RNA"]]$counts )
使用TransferData函数
在前面的方法学:使用singleR基于自建数据库来自动化注释单细胞转录组亚群,我们拿到了 sce.singleR.Rdata 文件里面是一个已经降维聚类分群并且注释好的Seurat对象。这个文件会很小,因为细胞数量确实是不多,但是已经是有 fibro 和endo以及周细胞和SMC 信息,以及部分免疫细胞亚群信息。
核心流程就是 FindTransferAnchors 和 TransferData 函数,即可把两个Seurat对象关联起来,其中一个Seurat里面是有单细胞亚群注释信息,就可以迁移到另外一个对象里面,如下所示:
load(file = 'singleR/sce.singleR.Rdata')
sce.singleR
table(sce.singleR$paper)
sce.ref = sce.singleR
source('scRNA_scripts/lib.R')
sce.all.int = readRDS('2-harmony/sce.all_int.rds')
sce.query = sce.all.int
sce.anchors <- FindTransferAnchors(reference = sce.ref,
query = sce.query, dims = 1:30,
reference.reduction = "pca")
predictions <- TransferData(anchorset = sce.anchors,
refdata = sce.ref$paper, dims = 1:30)
sce.query <- AddMetaData(sce.query, metadata = predictions)
table(sce.query$predicted.id )
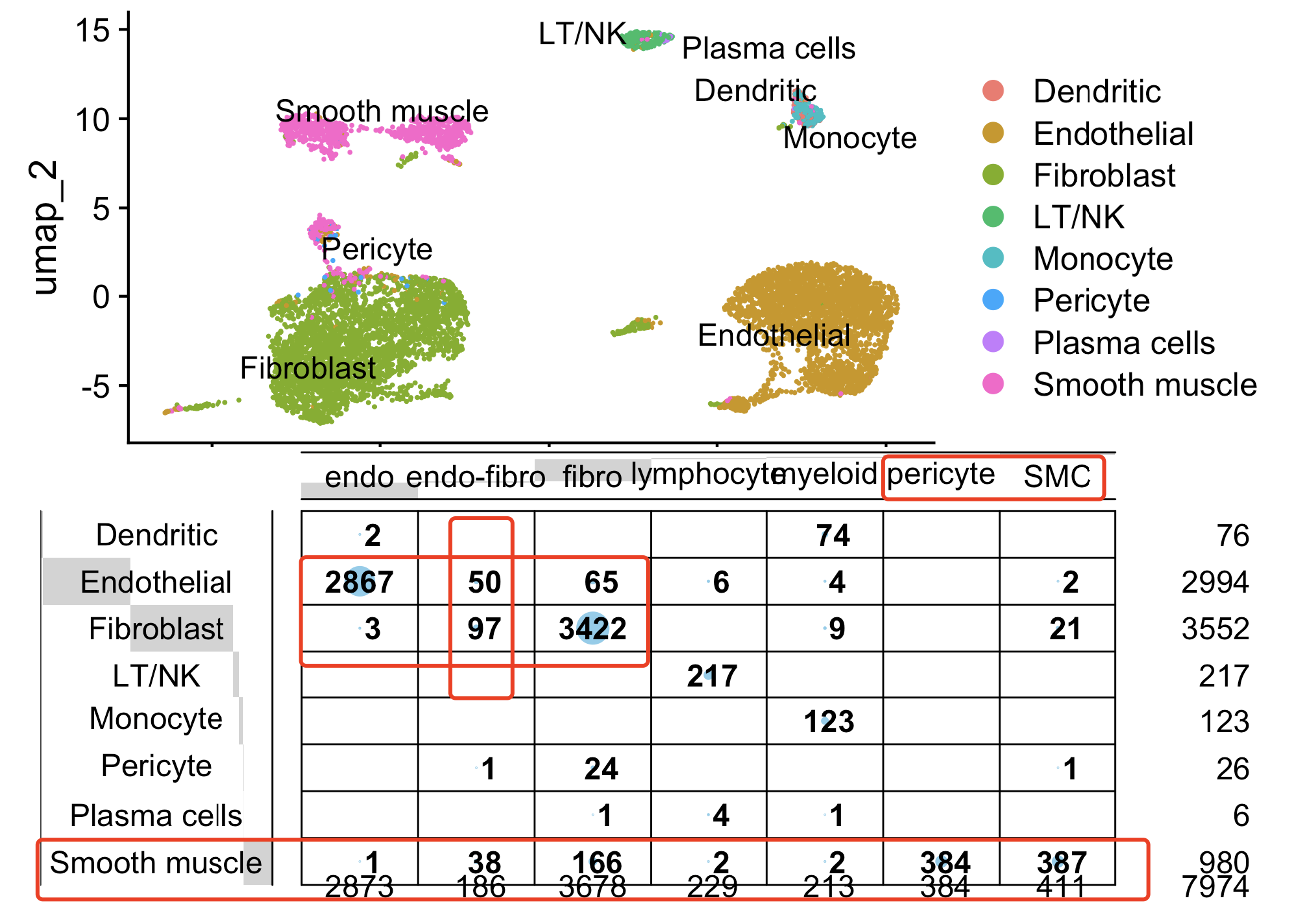
DimPlot(sce.query,group.by = 'predicted.id',label = T,repel = F)
load('phe.Rdata')
gplots::balloonplot(table(sce.query$predicted.id,phe$celltype ))
可以看到的是,从效果的角度来说,跟前面我们推荐了方法学:使用singleR基于自建数据库来自动化注释单细胞转录组亚群,对比,不知道为什么Seurat的TransferData函数没办法区分SMC和周细胞,其实在UMAP图是还是可以看到清晰可见的界限。前面的singleR就没有遇到这个问题: