寒假期间安排了一些小伙伴练习单细胞,其中一个文献是:《A comprehensive single-cell map of T cell exhaustion-associated immune environ- ments in human breast cancer》,它配套的数据在 E-MTAB-10607 可以看到,但是小伙伴在降维聚类分群的时候实在是没办法达到原文的漂亮的结果:

文献里面提到了是标准的商业化的10x技术的单细胞转录组,After standard data pre-processing, 119,000 high-quality cell measurements remained in the dataset
第一层次降维聚类分群很清晰:
- EPCAM/CDH1 for epithelial cells,
- CD3/CD4/CD8/NCR1 for the T and NK cell fraction,
- CD14/ITGAX/HLA- DRA for myeloid cells,
- PECAM1/VWF for endothelial cells,
- PDGFRB/FAP for fibroblasts,
- MS4A2 for mast cells and basophils,
- MS4A1 for B cells,
- Ig-encoding transcripts for plasma cells
让我们一起来看看小伙伴们的复现情况吧。首先是下载表达量矩阵的txt文件,如下所示的方法:
# 在Linux的wget命令可以得到网页文件:index.html
wget https://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/
# 从网页文件中提取count matrix 文件名字
grep href index.html |sed 's/.*href="//;s/".*//'|grep txt|grep count_matrix >count_matrix.list
cat count_matrix.list |while read a;do echo "wget -c ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/$a";done >run.sh
bash run.sh
其中这个 run.sh 的脚本内容如下所示 :
wget -c ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB011_singlecell_count_matrix.txt
wget -c ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB035_singlecell_count_matrix.txt
wget -c ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB075_singlecell_count_matrix.txt
wget -c ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB102_singlecell_count_matrix.txt
wget -c ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB111_singlecell_count_matrix.txt
wget -c ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB129_singlecell_count_matrix.txt
wget -c ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB165_singlecell_count_matrix.txt
wget -c ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB171_singlecell_count_matrix.txt
wget -c ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB184_singlecell_count_matrix.txt
wget -c ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB212_singlecell_count_matrix.txt
wget -c ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB214_singlecell_count_matrix.txt
wget -c ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB226_singlecell_count_matrix.txt
wget -c ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB330_singlecell_count_matrix.txt
wget -c ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB338_singlecell_count_matrix.txt
很简单的就可以下载这些txt文件啦;
ls -lh *txt |cut -d" " -f 5-
529M 2月 15 12:14 TBB011_singlecell_count_matrix.txt
488M 2月 15 12:15 TBB035_singlecell_count_matrix.txt
418M 2月 15 12:15 TBB075_singlecell_count_matrix.txt
295M 2月 15 12:16 TBB102_singlecell_count_matrix.txt
343M 2月 15 12:16 TBB111_singlecell_count_matrix.txt
799M 2月 15 12:17 TBB129_singlecell_count_matrix.txt
331M 2月 15 12:18 TBB165_singlecell_count_matrix.txt
422M 2月 15 12:18 TBB171_singlecell_count_matrix.txt
612M 2月 15 12:19 TBB184_singlecell_count_matrix.txt
393M 2月 15 12:19 TBB212_singlecell_count_matrix.txt
497M 2月 15 12:20 TBB214_singlecell_count_matrix.txt
428M 2月 15 12:21 TBB226_singlecell_count_matrix.txt
435M 2月 15 12:21 TBB330_singlecell_count_matrix.txt
730M 2月 15 12:22 TBB338_singlecell_count_matrix.txt
其实可以看到这些链接是有规律的,每个样品有表达量矩阵(singlecell_count_matrix.txt),也有配套的细胞注释信息文件(complete_singlecell_metadata.txt),其实可以简单的构建 shell 脚本如下所示:
#!/bin/bash
for i in {011,035,075,102,111,129,165,171,184,212,214,226,330,338}; do
wget -c "ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB${i}_singlecell_count_matrix.txt"
done
for i in {011,035,075,102,111,129,165,171,184,212,214,226,330,338}; do
wget -c "ftp://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/607/E-MTAB-10607/Files/TBB${i}_complete_singlecell_metadata.txt"
done
如下所示:
wc -l *
12496 TBB011_complete_singlecell_metadata.txt
11381 TBB035_complete_singlecell_metadata.txt
9902 TBB075_complete_singlecell_metadata.txt
6959 TBB102_complete_singlecell_metadata.txt
8074 TBB111_complete_singlecell_metadata.txt
18963 TBB129_complete_singlecell_metadata.txt
7802 TBB165_complete_singlecell_metadata.txt
9997 TBB171_complete_singlecell_metadata.txt
13010 TBB184_complete_singlecell_metadata.txt
9283 TBB212_complete_singlecell_metadata.txt
11730 TBB214_complete_singlecell_metadata.txt
10129 TBB226_complete_singlecell_metadata.txt
9811 TBB330_complete_singlecell_metadata.txt
17248 TBB338_complete_singlecell_metadata.txt
读取表达量矩阵并且降维聚类分群
很简单的一个循环即可哈,们从2024开始的教程都是基于Seurat的V5版本啦,之前已经演示了如何读取不同格式的单细胞转录组数据文件,如下所示:
这个文献的配套的数据在 E-MTAB-10607 ,是每个样品一个独立的txt文件,所以如下所示的读取方式即可:
rm(list=ls())
options(stringsAsFactors = F)
source('scRNA_scripts/lib.R')
getwd()
###### step1: 导入数据 ######
# 参考:https://mp.weixin.qq.com/s/tw7lygmGDAbpzMTx57VvFw
dir='inputs/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[7]
print(pro)
ct <- data.table::fread( file.path(dir,pro),
data.table = F)
ct[1:4,1:4]
rownames(ct)=ct[,1]
ct=ct[,-1]
sce =CreateSeuratObject(counts = ct ,
project = gsub('_singlecell_count_matrix.txt.gz','',pro) ,
min.cells = 5,
min.features = 300 )
return(sce)
})
do.call(rbind,lapply(sceList, dim))
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples )
sce.all <- JoinLayers(sce.all)
后面只需要针对这个 sce.all 变量里面的Seurat对象进行标准的降维聚类分群即可,可以看到如果超级低的分辨率情况下,已经算是比较清晰的分群了,唯一麻烦的就是0群里面很明显就是有一点点的b细胞混入,因为他们都是淋巴系的免疫细胞,聚在一起很正常的!
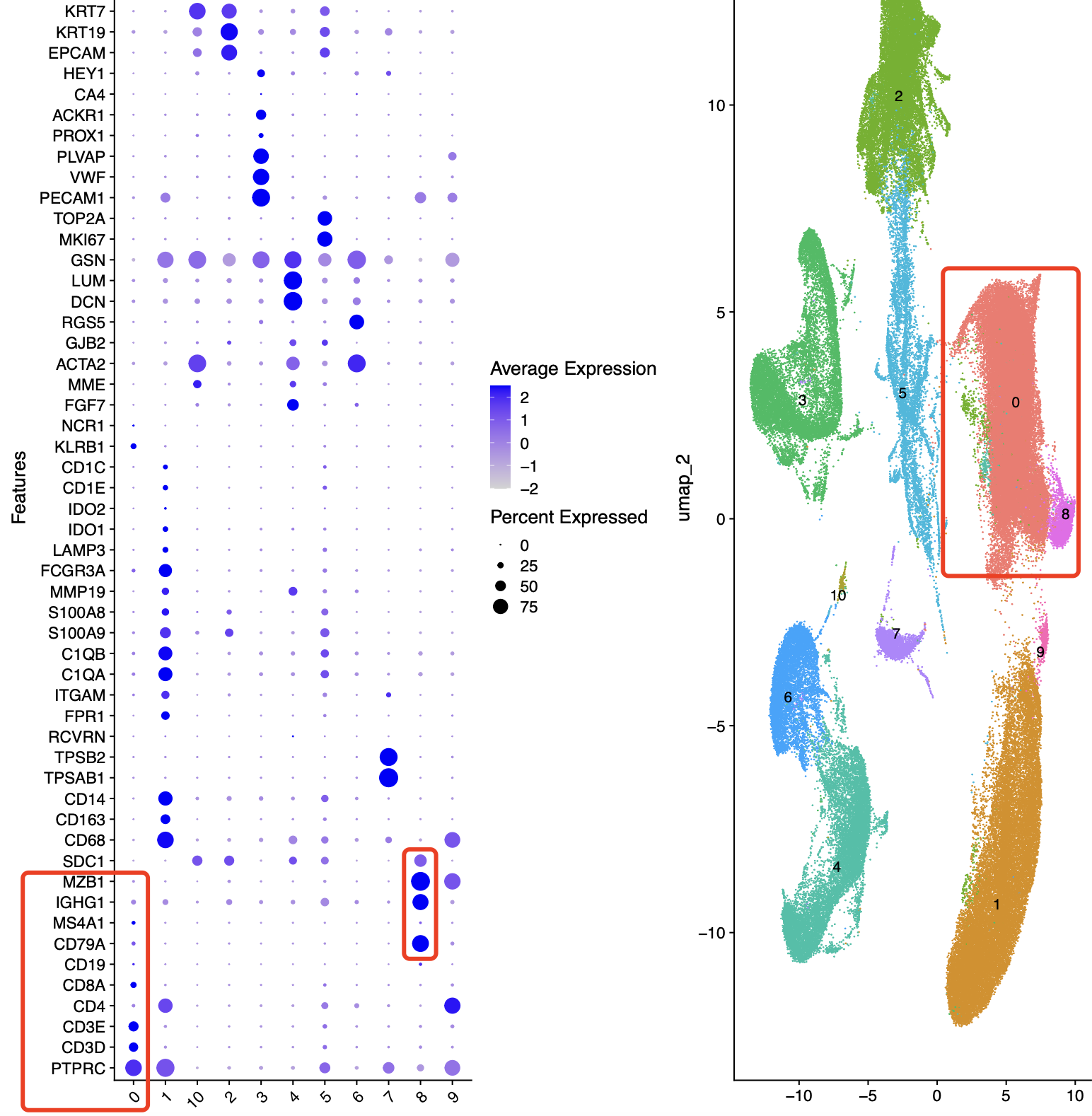
我们可以简简单单的提高一点分辨率,就可以看到b淋巴细胞会跟t淋巴细胞有一点点界限了,如下所示:
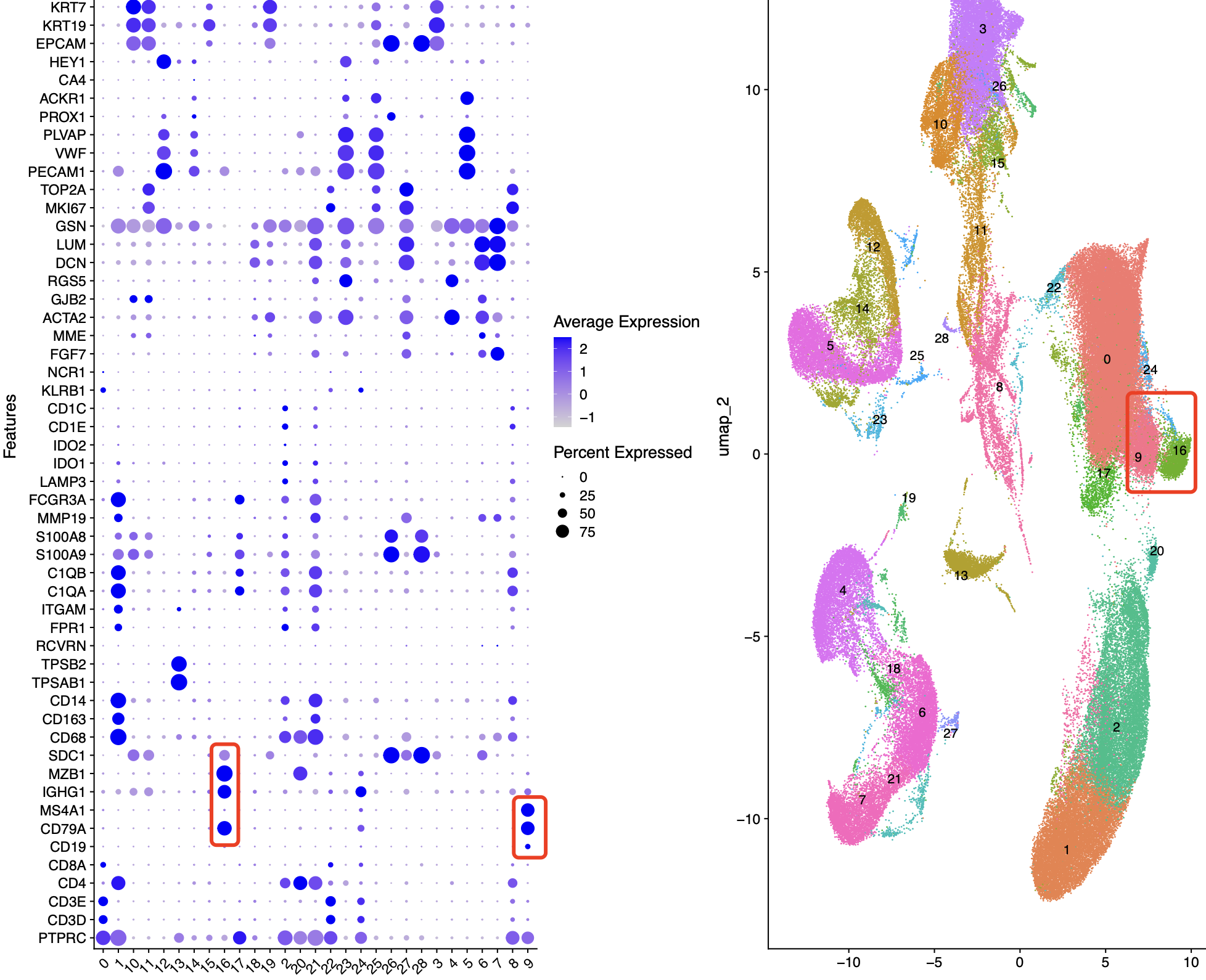
但是很明显,这个降维聚类分群其实是跟原文作者的漂亮的结果是有差距的。原文的t淋巴细胞跟b淋巴细胞并没有任何的混杂。那么,我们的标准代码问题出在哪里呢?因为 作者其实是给出来了自己的单细胞亚群的生物学注释信息,所以理论上我们读取每个样品的配套的细胞注释信息文件(complete_singlecell_metadata.txt)就可以跟自己的注释结果对比一下啦!
首先呢,毫无疑问,我们的结果确实是比较丑,如下所示:
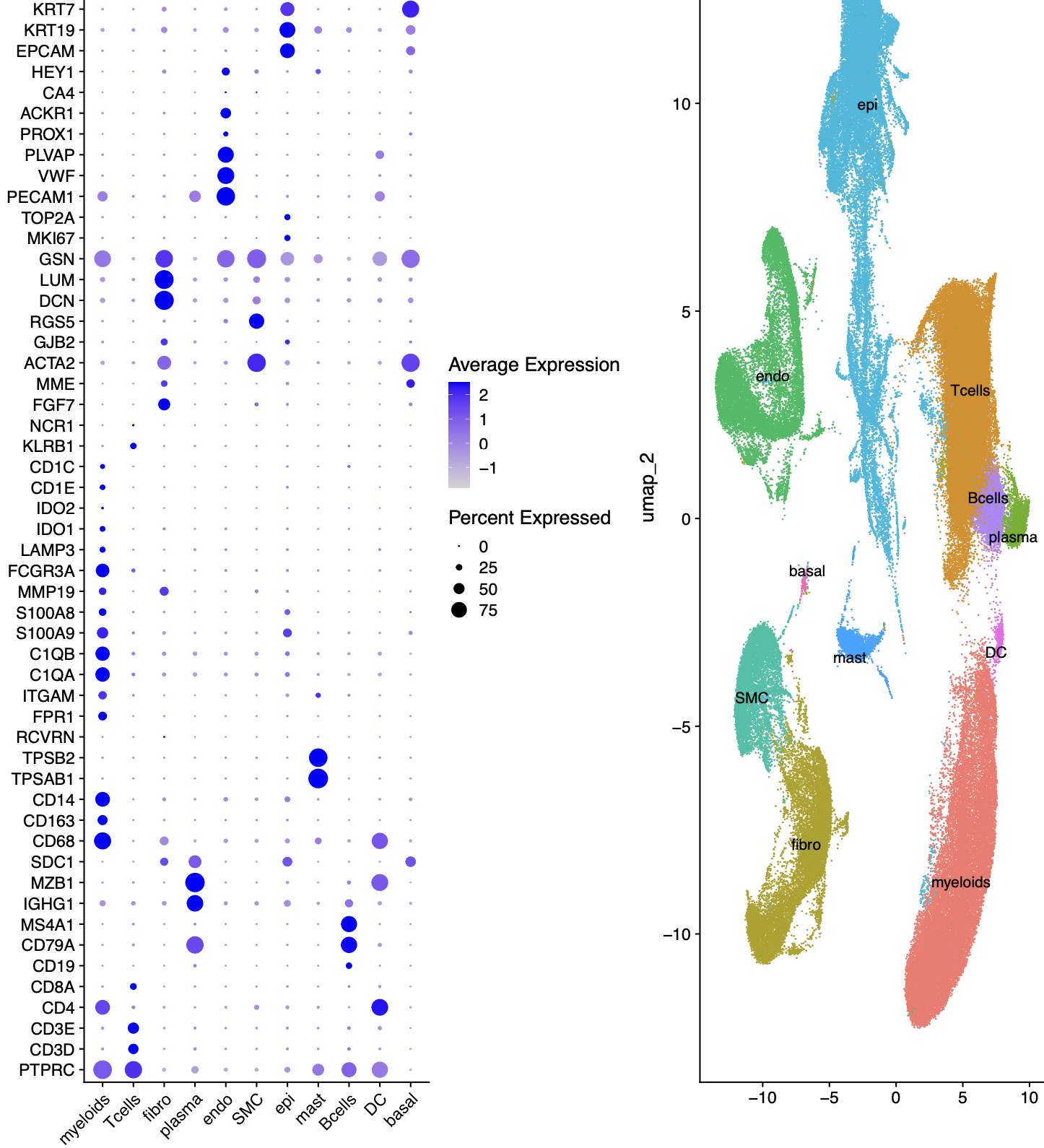
但是我们的结果合理性是没有问题的,因为这个是算法本身的限制,如果想要非常完美非常漂亮大家结果,这个单细胞转录组数据分析流程里面的降维聚类分群的每个步骤都需要大量的调整参数,工作量实在是太大了,而且我们完全是可以验证它的必要性是否强!
我们的图虽然丑爆了,但是只需要它的降维聚类分群后的单细胞亚群的生物学名字是ok的,就不怕,因为我们做单细胞转录组数据分析的核心是给每个细胞一个合理的身份,而不是“屎上雕花”让这个umap或者tSNE图多好看!!!!
比对两次分群后的单细胞名字
首先看看作者的:
dir='meta/'
samples=list.files( dir )
samples
ctList = lapply(samples,function(pro){
# pro=samples[7]
print(pro)
ct <- data.table::fread( file.path(dir,pro),
data.table = F)
ct[1:4,1:4]
return(ct)
})
do.call(rbind,lapply(ctList, dim))
phe2 = do.call(rbind,ctList)
table(phe2$cell_type)
load(file = 'phe.Rdata')
table(phe$celltype)
可以看到其实大家的降维聚类分群后的单细胞亚群结果确实是有一点点区别的,如下所示:
> table(phe2$cell_type)
B cell endothelial epithelial fibroblast granulocyte myeloid pDC
4737 10397 18241 16704 2684 26651 770
plasma cell T/NK cell
2477 36146
> table(phe$celltype)
basal Bcells DC endo epi fibro mast myeloids plasma
216 4071 615 12417 25698 12260 2897 25577 2091
SMC Tcells
6770 35164
> dim(phe);
[1] 127776 16
> dim(phe2);
[1] 156771 10
但是这个区别是否能够被容忍呢?一个简简单单的可视化,就能看看两次结果的交集(127776个细胞)如何,如下所示:
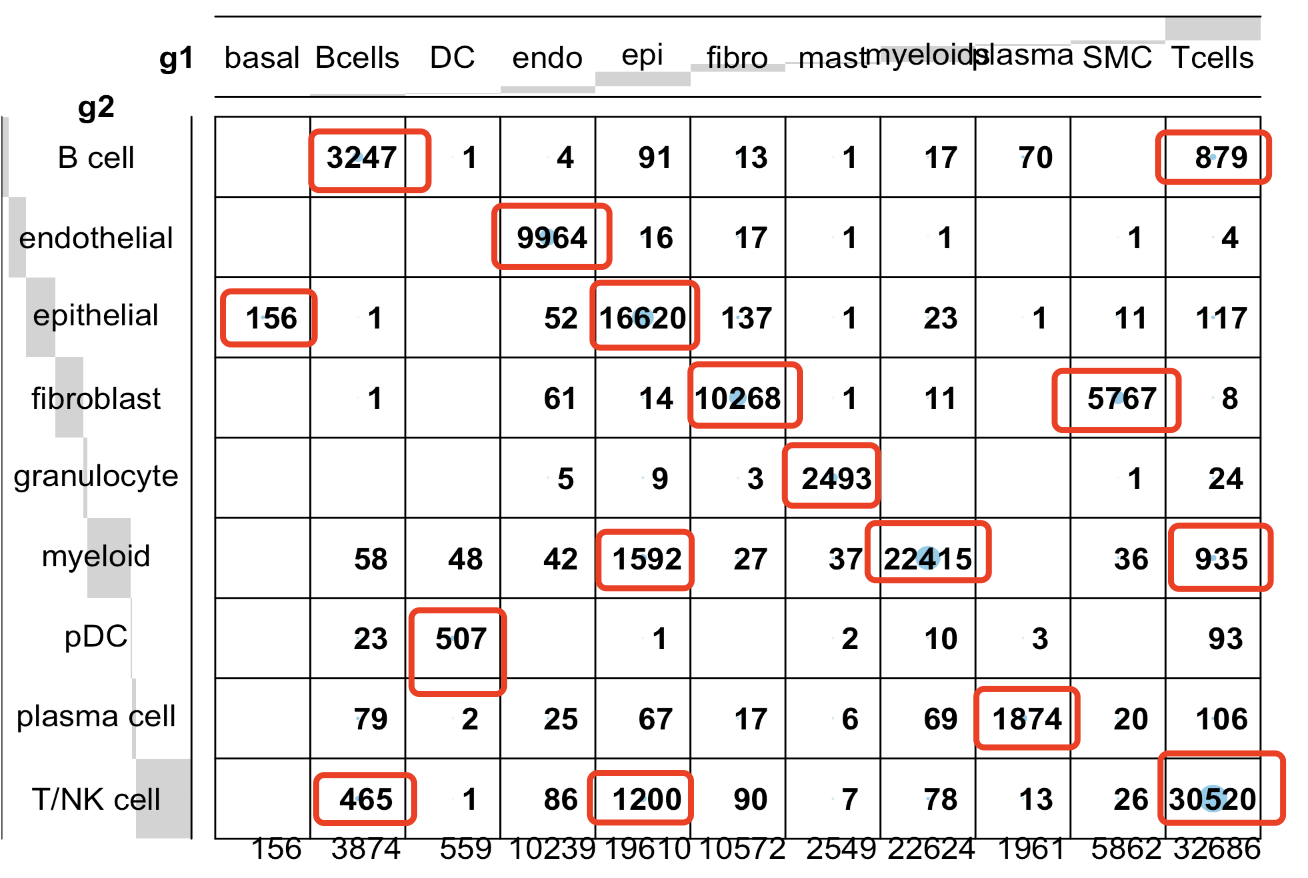
可以看到,我们的命名系统里面可以区分出来成纤维里面的SMC,这个被作者选择性忽略,同样的我们区分出来了basal这个上皮细胞,也是被作者直接混入到上皮细胞里面了。这个并没有什么冲突,仅仅是因为大家的目标不一样,比如我们可以说广东广西人是不一样的,也可以统称为两广人。
然后,让我们比较难抉择的地方就是b淋巴细胞会跟t淋巴细胞的混杂问题,虽然说作者的结果是漂亮的,但是实际上很难说它的结果是正确的,漂亮并不等于正确,其实这个时候甚至是可以有一个课题了,来探索假阳性和假阳性问题。那个465个细胞群体被作者认为是t细胞但是我认为是b细胞,而另外的879个细胞群体被我认为是t细胞但是作者认为是b细胞,孰真孰假现在很难肉眼看出来。
其实本质上是单细胞数据质量问题,如果是严格的过滤,比如文章提到的可以低于12万的细胞数量。我猜测,无论是怎么样的过滤或者调参,其实仍然是有一些髓系免疫细胞和上皮细胞混入到t淋巴系细胞大亚群里面,或者各种混入,但是它们无伤大雅的,因为我们还会进行第二层次的降维聚类分群啊,到时候再明确它的身份也不晚的!

我们的《标记基因》专辑目前主要是介绍了肿瘤相关单细胞转录组的第一层次降维聚类分群后的细分亚群:
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibo or CD31+,PECAM1,endo)
其中上皮细胞我们介绍了不同器官:
免疫细胞呢,主要是包括淋巴系(T,B,NK细胞)和髓系(单核,树突,巨噬,粒细胞)的两大类作为第二次细分亚群,但是目前我们才介绍到:
stromal 里面的fibo 和endo,我们就介绍了内皮细胞细分亚群,其实还有周细胞和平滑肌细胞,我们后期再慢慢补充。