我们粗浅的认为空间单细胞约等于10x技术,虽然说确实是有很多其它空间单细胞技术可以产出各式各样的数据。但是前期我们还是着重于介绍10x的空间单细胞文件格式详解 ,里面 提到的几个案例其实并不是所有的数据集都是可以分析的,因为作者提供的文件本身就是缺胳膊断腿的。
可以成功读取的单细胞空间转录组数据文件
比如2021-GSE158328-肠道发育的,自己下载 GSM4797916_A1.tar.gz ,然后解压可以看到它每个样品其实有两个文件夹 :
% ls -lh *|cut -d" " -f 7-
raw_feature_bc_matrix:
28K 2 20 2020 barcodes.tsv.gz
298K 2 20 2020 features.tsv.gz
18M 2 20 2020 matrix.mtx.gz
spatial:
1.6M 2 20 2020 aligned_fiducials.jpg
1.8M 2 20 2020 detected_tissue_image.jpg
163B 2 20 2020 scalefactors_json.json
4.6M 2 20 2020 tissue_hires_image.png
496K 2 20 2020 tissue_lowres_image.png
180K 2 20 2020 tissue_positions_list.csv
上面的两个文件夹需要独立读取,代码如下所示:
library(Seurat)
## 首先读取表达矩阵数据
ct = Read10X("./GSE158328_RAW/A1/raw_feature_bc_matrix")
## 然后读取image信息
img <- Read10X_Image(image.dir = file.path("./GSE158328_RAW/A1/",
"spatial"), filter.matrix = TRUE)
## 接着创建seurat对象
# https://rdrr.io/github/satijalab/seurat/src/R/preprocessing.R
sceP <- CreateSeuratObject(counts = ct, assay = "Spatial")
img <- img[Cells(x = sceP)]
DefaultAssay(sceP = img) <- "Spatial"
sceP[['slice1']] <- img
接着我们一起看看 2022-复旦高强课题组-结直肠癌肝转移空间单细胞数据:
# 空间单细胞的文件在文献pdf里面的可以找到地址:ST-colon2
% ls -lh *|cut -d" " -f 7-
77K 12 15 2021 barcodes.tsv
162K 12 15 2021 coordinates.tsv
15M 12 19 2021 filtered_feature_bc_matrix.h5
601K 12 15 2021 genes.tsv
127M 12 15 2021 matrix.mtx
166B 12 19 2021 scalefactors_json.json
16M 12 19 2021 tissue_hires_image.png
1.4M 12 19 2021 tissue_lowres_image.png
184K 12 19 2021 tissue_positions_list.csv
这个文献对每个样品提供了Market Exchange Format (MEX) 和 Hierarchical Data Format (HDF5) 两种格式的表达量矩阵,这个时候我们一般来说会选择那个 filtered_feature_bc_matrix.h5 读取后分析即可。但是需要简单的制作一下文件夹目录结构如下所示:
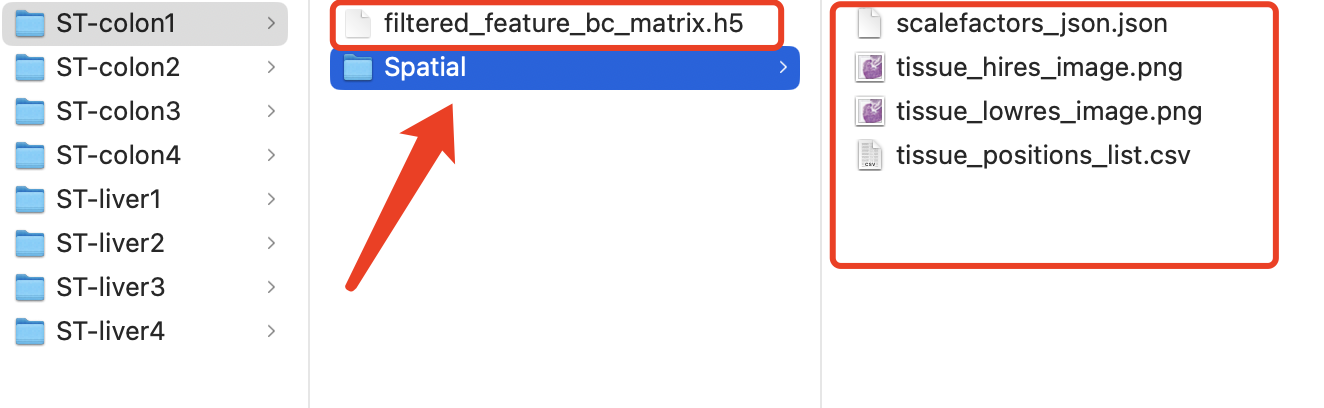
ST-colon1 % tree -h
[ 288] .
├── [ 192] Spatial
│ ├── [ 165] scalefactors_json.json
│ ├── [ 16M] tissue_hires_image.png
│ ├── [1.5M] tissue_lowres_image.png
│ └── [184K] tissue_positions_list.csv
├── [ 23M] filtered_feature_bc_matrix.h5
很清晰的,每个样品都是一个独立的文件夹,里面有一个h5文件,以及图片文件夹(Spatial),如下所示:
因为实在是太标准,所以读取它就一句话代码即可:
library(Seurat)
sceP = Load10X_Spatial(data.dir = "./inputs/ST-colon1/",
filename = "filtered_feature_bc_matrix.h5",
assay = "Spatial",
slice = "test")
理想是美好的,现实是骨感的
前面介绍10x的空间单细胞文件格式详解 的时候我们列出来了很多案例,其实更多的是没办法成功读取的, 比如2022的文章:《Different Molecular Features of Epithelioid and Giant Cells in Foreign Body Reaction Identified by Single-Cell RNA Sequencing》,给出来的空间单细胞文件是:
% ls -lh *|cut -d" " -f 7-
211M 5 17 2022 GSM6160936_vis01_Sponge-4.tif.gz
96K 5 13 2022 GSM6160936_visium_meta_data.txt.gz
6.6M 5 13 2022 GSM6160936_visium_raw_counts.txt.gz
可以看到,虽然说我们可以读取GSM6160936_visium_raw_counts.txt.gz去构建单细胞表达量矩阵对象,也可以读取GSM6160936_visium_meta_data.txt.gz看看具体的每个空间spot是什么细胞类型,它的属性,但是我们没办法读取 GSM6160936_vis01_Sponge-4.tif.gz这样的空间切片的图片信息。相当于就前功尽弃了,但是考虑到它有tif.gz这样的空间切片的图片信息,其实是可以从fastq文件开始, 走spaceranger定量流程,详情请见:空间单细胞的上游定量流程(spaceranger,10x技术)
学徒作业
在 PRJNA791135-SRA Links for BioProject (Select 791135) - SRA - NCBI 可以看到这个空间单细胞转录组的fastq文件,下载后,配套它的tif.gz这样的空间切片的图片走spaceranger定量流程,然后简单的处理它!做一个初步的降维聚类分群即可哈:
