fastqc软件的使用
一:下载安装该软件
具体搜索其地址下载,fastqc是一个java软件,下载后可以直接使用,但是需要自行配置好java环境,具体配置方法,见linux下java配置。
二:准备数据
数据就是我们测序得到的fastq文件的reads,压缩包也可以直接运行
三:运行命令
我习惯了批处理解决问题,脚本如下:
for id in *fastq
do
echo $id
/home/jmzeng/bio-soft/FastQC/fastqc $id
Done
运行过程中会显示以下的提示信息
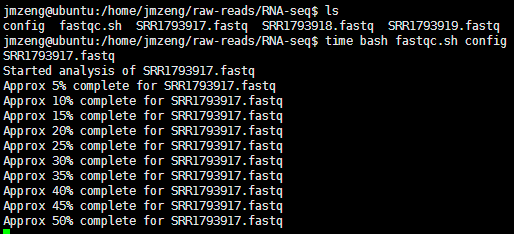
估计还是要运行很久的,比较这几个RNA-seq文件每个都是16G的
按住ctrl+A+D即可退出该后台,继续去前台执行简单任务
好像二十分钟就跑完了
![]() 输出文件如下
输出文件如下
四:输出文件解读
可以直接打开那个html网页文件就可以查看每一个图片内容,也可以解压那个zip压缩包具体看每一张图片
下载fastqc跑出来的结果一个个解读
1,简单统计表格
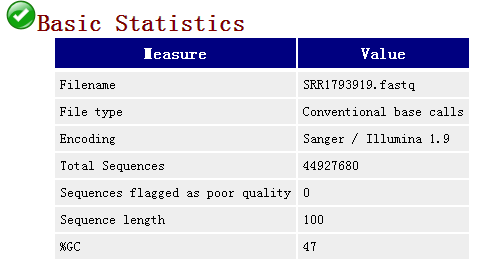
这些英文我就不翻译了,reads均长是100bp,共四千多万条reads
2,测序质量图
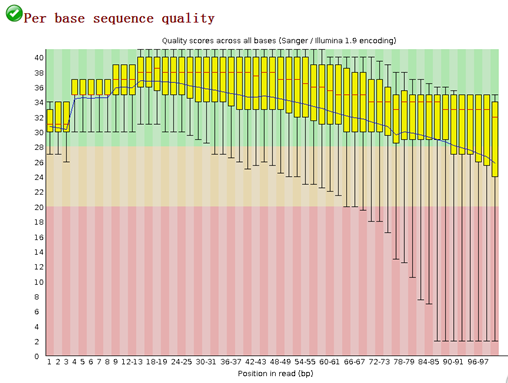
这个图其实很容易看,就是100bp长度reads上的1-100的坐标在这四千万条reads里面的测序质量的箱线图,看那个红线均值就可以了,超过Q30就蛮好了,超过Q20也是合格的
3,碱基(A,T,C,G)含量图
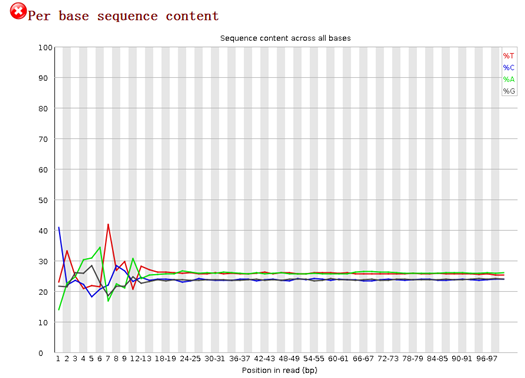
这也是100bp长度reads上的1-100的坐标在这四千万条reads里面的A,T,C,G的比例,如果是全基因组全转录组的随机打断,那么就应该A,T,C,G的比例都接近于25%,如果测序是有目的性的,那么比例也就相应的改变了
4,reads的GC含量频数分布图
这是对四千万条reads里面的GC含量值做统计密度曲线,可以看到绝大部分的reads的GC含量都集中在50%附近。极端情况很少。
5,reads长度分布图
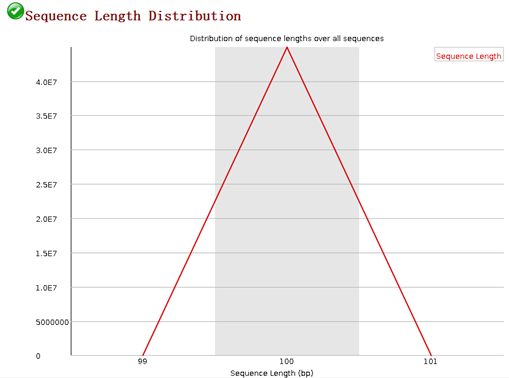
可以看到大多reads都是100bp长度,很整齐
6,可能的重复序列表格
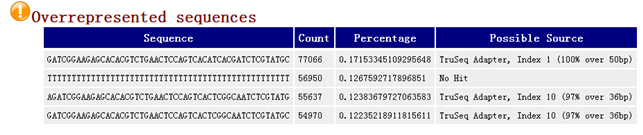
可以看到这些重复序列比例很高,高达千分之一,而且被注释了可能的来源,adapter,是需要去除的。
好样的