我看到了《肿瘤资讯》公众号发布了浙江省肿瘤医院泌尿外科副主任医师徐一鹏的《从模型出发看肿瘤药物研究》观点,里面提到了肿瘤细胞系,即Cell lines的两个缺点:
- 首先是是不具备肿瘤微环境。虽然细胞株都是从病人的肿瘤组织中获取,但是在细胞株创建伊始就要进行纯化,随着不断地体外增殖,尽管该肿瘤细胞株中还含有肿瘤细胞,但在纯化过程中失去了肿瘤所具备的微环境,例如支持性的非肿瘤细胞的基质、血液系统、免疫系统,以及一些其它的肿瘤微环境因子。
- 其次是不断的培养和传代过程对于细胞株而言是一个筛选的过程,一些优势细胞会逐步留存。而且随着体外培养环境的改变,细胞会逐步适应体外培养环境,而失去原有特征。2018年发表在Nature杂志的一篇文献说明了肿瘤细胞系的缺陷。该研究对两个实验室培养的 106 个人类细胞系进行了基因组分析。研究结果出乎意料地发现来源于不同实验室的同种细胞株所表现的表型是不同的。其
本来呢,大家期待肿瘤类器官(PDO)作为肿瘤模型的革命性技术是可以解决上面的两个问题。首先呢,肿瘤类器官优势在于它源自各个病人的原始肿瘤组织,并且基于其原代培养的特性,其与原始肿瘤组织在病理特征、基因突变等多维度具有一致性,可再现肿瘤异质性,并且可传代、冻存,还可进行基因操作。但是呢, 早期的肿瘤类器官其实也是没有肿瘤微环境的,仅仅是恶性肿瘤上皮细胞被培养。
比如我们前面的单细胞转录组数据分析结果,就可以看到,类器官里面只有上皮细胞,而肿瘤病人的样品里面就有微环境的,这个降维聚类分群详见:有了单细胞数据的类器官研究其实可以不做常规基因组和转录组啦
上面的上皮细胞就是恶性的肿瘤细胞,是通过inferCNV算法判定的,详见:有了单细胞数据的类器官研究其实可以不做常规基因组和转录组啦。
也就是说,类器官培养物确实是很好的维持了原位肿瘤的遗传突变特性,但是它的缺点就是丢失了肿瘤微环境,因为培养物里面的都是恶性的肿瘤上皮细胞。不过,上面的案例毕竟是就一个病人培养前后,因为单细胞研究毕竟是比较烧钱,很难做出大量的样品,单个样品里面的信息很难被归纳整理成为客观规律。这个时候,我们可以使用去卷积算法:贝叶斯棱镜(BayesPrism)来针对传统的bulk转录组数据去推测里面的不同单细胞亚群组成比例情况,当然了这个过程是需要一个特定肿瘤单细胞数据集来作为参考,然后还得挑选合适的肿瘤类器官bulk转录组表达量矩阵。
第一步:构建做参考的单细胞亚群数据集
首先,我们必须提前做好了单细胞转录组数据集的降维聚类分群和命名,这个就是走常规的单细胞转录组降维聚类分群代码,可以看 链接: https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh ,基本上大家只需要读入表达量矩阵文件到r里面就可以使用Seurat包做全部的流程。
然后就可以处理单细胞结果成为贝叶斯棱镜(BayesPrism)输入文件,代码如下所示:
rm(list = ls())
library(ggplot2)
library(dplyr)
library(gridExtra)
library(grid)
#pacman::p_install_gh("Danko-Lab/BayesPrism/BayesPrism")
library(BayesPrism)
library(Seurat)
library(ggplot2)
library(clustree)
library(cowplot)
library(scater)
sce=readRDS( "2-harmony/sce.all_int.rds")
load('phe.Rdata')
Idents(sce) = phe$celltype
table(Idents(sce))
# 可以针对这个单细胞对象,取子集
sce = subset(sce,downsample=100)
table(Idents(sce))
library(scater)
sub.sce <- SingleCellExperiment(
assays = list(counts = sce@assays$RNA$counts),
colData = data.frame(cellType=Idents(sce),sampleID=colnames(sce))
)
sub.sce
colData(sub.sce)
unique(sub.sce$cellType)
save(sub.sce,file = 'sub.sce_for_bayesPrism.Rdata')
其实上面的代码基本上没有什么特殊的操作,就是把Seurat包的单细胞对象变成了scater包的对象, 这个是美国纽约的科研课题组和英国那边的课题组的学术斗争。
第二步:然后处理传统的bulk转录组表达量矩阵
我们这里选择数据集:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE84073
对应的文章是:uman primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat Med 2017 Dec;23(12):1424-1435. PMID: 29131160
可以看到这个研究里面的转录组测序实验设计还蛮复杂的,包括3种肝癌human primary liver cancer (PLC),就是different tumor subtypes (HCC, CHC, CC),以及正常的肝组织样品,还有他们的类器官培养物:
GSM2653819_Counts_notmergedTR_Healthy1_Tissue_1.txt.gz
GSM2653820_Counts_notmergedTR_Healthy1_Tissue_2.txt.gz
GSM2653821_Counts_notmergedTR_Healthy2_Tissue.txt.gz
GSM2653822_Counts_notmergedTR_Healthy3_Tissue.txt.gz
GSM2653833_Counts_notmergedTR_Healthy1_Organoid_1.txt.gz
GSM2653834_Counts_notmergedTR_Healthy1_Organoid_2.txt.gz
GSM2653835_Counts_notmergedTR_Healthy2_Organoid.txt.gz
GSM2653837_Counts_notmergedTR_Healthy3_Organoid.txt.gz
详细的样品分布如下所示:
> table(colD)
cancer
group CC CHC HCC Healthy
Organoid 7 5 3 6
Tissue 4 3 3 4
需要自己下载 GSE84073_RAW.tar文件然后解压后存放在countsFiles文件夹里面,然后批量读取的 具体的代码是:
rm(list = ls())
options(stringsAsFactors = F)
fs=list.files('countsFiles/')
a=do.call(cbind,lapply(list.files('countsFiles/'), function(x){
read.table(file.path('countsFiles',x),
header = T,sep = '\t',row.names = 1)[,2]
}))
a[1:4,1:4]
rownames(a)=rownames(read.table( file.path('countsFiles',fs[2]),
header = T,sep = '\t',row.names = 1))
colnames(a)=gsub('.txt.gz','',substring(fs,nchar("GSM2653853_Counts_notmergedTR_")+1,1000))
exprSet=a
boxplot(log2(edgeR::cpm(exprSet)+1),las=2)
print(dim(exprSet))
exprSet=exprSet[apply(exprSet,1,
function(x) sum(x>1) > floor(ncol(exprSet)/20)),]
print(dim(exprSet))
ensembl_matrix =exprSet
library(AnnoProbe)
head(rownames(ensembl_matrix))
ids=annoGene(rownames(ensembl_matrix),'ENSEMBL','human')
head(ids)
ids=ids[!duplicated(ids$SYMBOL),]
ids=ids[!duplicated(ids$ENSEMBL),]
symbol_matrix= ensembl_matrix[match(ids$ENSEMBL,rownames(ensembl_matrix)),]
rownames(symbol_matrix) = ids$SYMBOL
symbol_matrix[1:4,1:4]
library(stringr)
colnames(exprSet)
cancer=gsub("\\d",'',str_split(colnames(exprSet),'_',simplify = T)[,1],perl = T)
table(cancer)
group_list=str_split(colnames(exprSet),'_',simplify = T)[,2]
table(group_list)
colD=data.frame(group=group_list,
cancer=cancer)
rownames(colD)=colnames(exprSet)
table(colD)
exprSet[1:4,1:4]
save(exprSet,symbol_matrix,colD,file = 'bulk_input.Rdata')
第三步:走贝叶斯棱镜算法
针对上面的bulk转录组测序后的表达量矩阵,以及肿瘤样品的单细胞转录组降维聚类分群结果文件,就可以走去卷积算法:贝叶斯棱镜(BayesPrism)。
首先是准备工作,就是上面的两个步骤的输出结果需要这个时候读取
rm(list = ls())
options('scipen'=100,'digits'=4)
library(ggplot2)
library(dplyr)
library(gridExtra)
library(grid)
library(BayesPrism)
library(Seurat)
library(ggplot2)
library(clustree)
library(cowplot)
library(scater)
load(file = 'sub.sce_for_bayesPrism.Rdata')
load('bulk_input.Rdata')
dir.create('run_BayesPrism/')
setwd('run_BayesPrism/')
bulk_matrix = t(as.matrix(symbol_matrix))
bulk_matrix[1:4,1:4]
sub.sce
colData(sub.sce)
unique(sub.sce$cellType)
single_cell_matrix <- t(as.matrix(assay(sub.sce)))
single_cell_matrix[1:4,1:4]
# Get cell type and cell state labels
cell_types <- sub.sce$cellType
sort(table(cell_types))
# BayesPrism deconvolution
set.seed(1234)
然后就可以运行:
if(!file.exists(file = 'prop_BayesPrism.Rdata')){
# Cleanup genes
sc.dat.filtered <- cleanup.genes(input = single_cell_matrix,
input.type = "count.matrix",
species="hs",
gene.group = c("Rb","Mrp","other_Rb","chrM","MALAT1"),
exp.cells = 5)
plot.bulk.vs.sc (sc.input =sc.dat.filtered,
bulk.input = bulk_matrix
#pdf.prefix="gbm.bk.vs.sc" specify pdf.prefix if need to output to pdf
)
# Filter to protein coding genes for faster computation
sc.dat.filtered.pc <- select.gene.type(sc.dat.filtered,
gene.type = "protein_coding")
myPrism <- new.prism(reference = sc.dat.filtered.pc,
mixture = bulk_matrix,
input.type = "count.matrix",
cell.type.labels = cell_types,
cell.state.labels = cell_types,
key= NULL , # no malignant cells
outlier.cut=0.01,
outlier.fraction=0.1,
)
# Run BayesPrism
bp.res <- run.prism(prism = myPrism, n.cores=6)
# Save BayesPrism object for later perusal
save(bp.res,file = 'prop_BayesPrism.Rdata')
}
load(file = 'prop_BayesPrism.Rdata')
prop_BayesPrism=get.fraction(bp = bp.res,
which.theta = "final",
state.or.type = "type")
rowSums(prop_BayesPrism)
colnames(prop_BayesPrism)
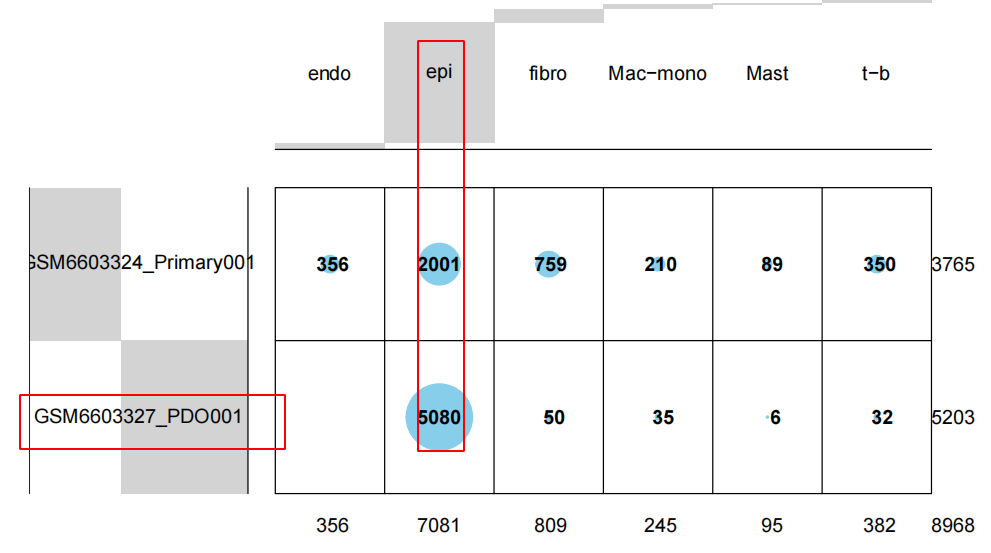
我们可以简单的可视化这个结果,首先可以看到基本上就是类器官培养物里面都是上皮细胞啦,都是接近于百分百的,但是肿瘤组织里面就有各种免疫微环境啦,上皮细胞比例也很低。
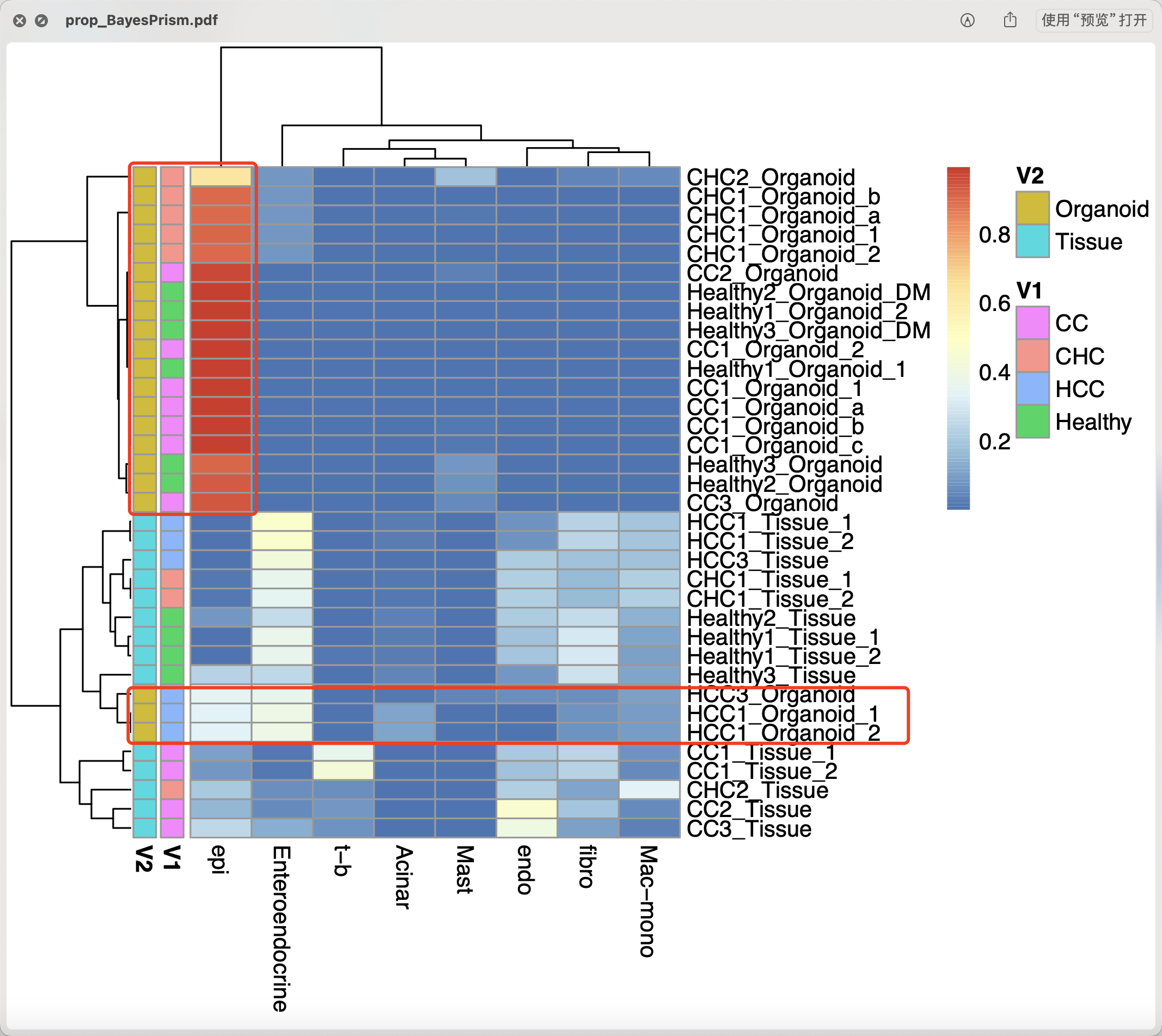
值得注意的是,不知道为什么有3个类器官培养物跟其它类器官距离有点远,不知道是不是它培养失败。或者说它克服了传统类器官培养会丢失肿瘤微环境的这个缺点?
当然了目前本来就是有一些类器官改良方法,比如结合3D打印去建造3D肿瘤模型。3D打印的最大优势在于可以让更多细胞,比如间质细胞、干细胞,以及巨噬细胞等存在于模型中。此外,还可以让细胞比例达到可控,可以在体外实现高度仿生的肿瘤免疫微环境。3D打印的创新点在于,从细胞角度而言,通过建立复杂、仿生的多细胞肿瘤3D模型,解决了难以在体外进行人源肿瘤免疫微环境研究的问题;从基质角度而言,通过调控生物材料属性,建立血管化的不同亚型肿瘤模型,开发体外还原肿瘤异质性、研究血管生成新方法。
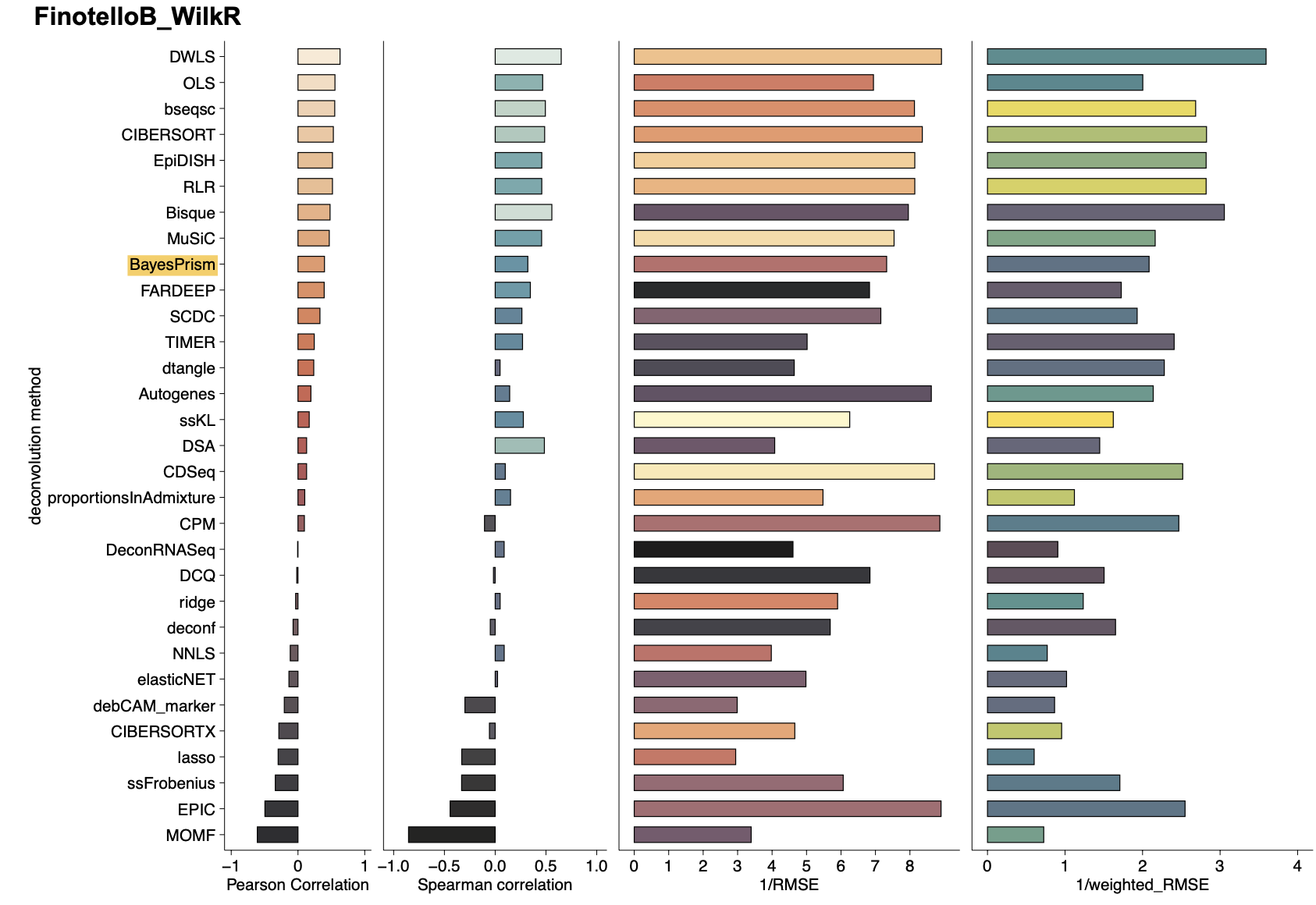
关于去卷积
上面我们演示的是卷积算法:贝叶斯棱镜(BayesPrism),跑一下需要半个小时左右,而且结果非常符合预期。其实能实现类似的需求的算法非常多,比如新鲜出炉的一个生物信息学软件测评文章:《CATD: a reproducible pipeline for selecting cell-type deconvolution methods across tissues》,就是对比了 31 deconvolution methods,主要的4个指标 (Pearson Correlation, Spearman Correlation, RMSE and weighted RMSE 的测评结果如下所示: