前面我们在 为什么做拟时序 提到了其实可以把拟时序分析简化成为了主要是为了展示差异细节,然后在 拟时序的正确姿势 我们3个案例来说明一下拟时序的正确姿势,错误示范,还有创新型的应用场景。
而且这些案例文献都不约而同的使用了monocle2这个软件,但是并不意味着它是金标准,也不意味着非monocle2不可。这个纯粹就是生物信息学领域的“马太效应”,大家都用monocle2做拟时序,所以后来者就简单的追随即可,而且绝大部分人其实并不关心算法细节,仅仅是为了做拟时序而做,那么就无所谓选择哪个软件了。实际上,做拟时序确实是有很多软件和算法的选择,我们可以来看看两个经典的综述,借此机会系统性梳理一下这方面背景知识。
首先看2016的综述对比10款算法软件
文章是:《Computational methods for trajectory inference from single-cell transcriptomics》,该综文章述对比10款算法软件并且列出来了各自的优缺点,可以看到那个时候monocle2做拟时序的突出就已经成定局了,而且它是引用率最高的!

总体上来说,这些软件首先区分成为了两个流派,就是专注于做Linear trajectories 的,以及可以做branched trajectories的。
然后它们这些软件算法都包含了两个步骤,就是降维(Dimensionality reduction)和轨迹构建(Trajectory modelling),这两个步骤的不同算法的组合就确定了不同的软件。比如最经典的monocle2做拟时序就是independent component analysis (ICA)的降维处理,配合 a minimal spanning tree (MST) 的构建拟时序关系算法。而其它软件会选择各种降维算法,比如 principal compo- nent analysis (PCA),diffusion maps, and t-SNE等等,或者各种其它构建拟时序关系算法。
然后看2019的综述对比45款算法软件
文章是:《A comparison of single-cell trajectory inference methods》工作量就更大了:we evaluated the accuracy, scalability, stability and usability of 45 TI methods 。
可以看到,前面的2016的综述还在使用Linear和branched分类,这次2019的综述就扩充到了7种不同轨迹形式:
- 循环(Cycle)
- 线性(Linear)
- 二分支(Bifurcation)
- 多分支(Multifurcation)
- 树状(Tree)
- 连通图(Connected graph)
- 不连通图(Disconnect graph)
并且是提出来了4个评价指标来测评这些软件工具: - HIM (Hamming-Ipsen-Mikhailov distance), F1 branches, F1 Milestones, and correlation metrics
如下所示:
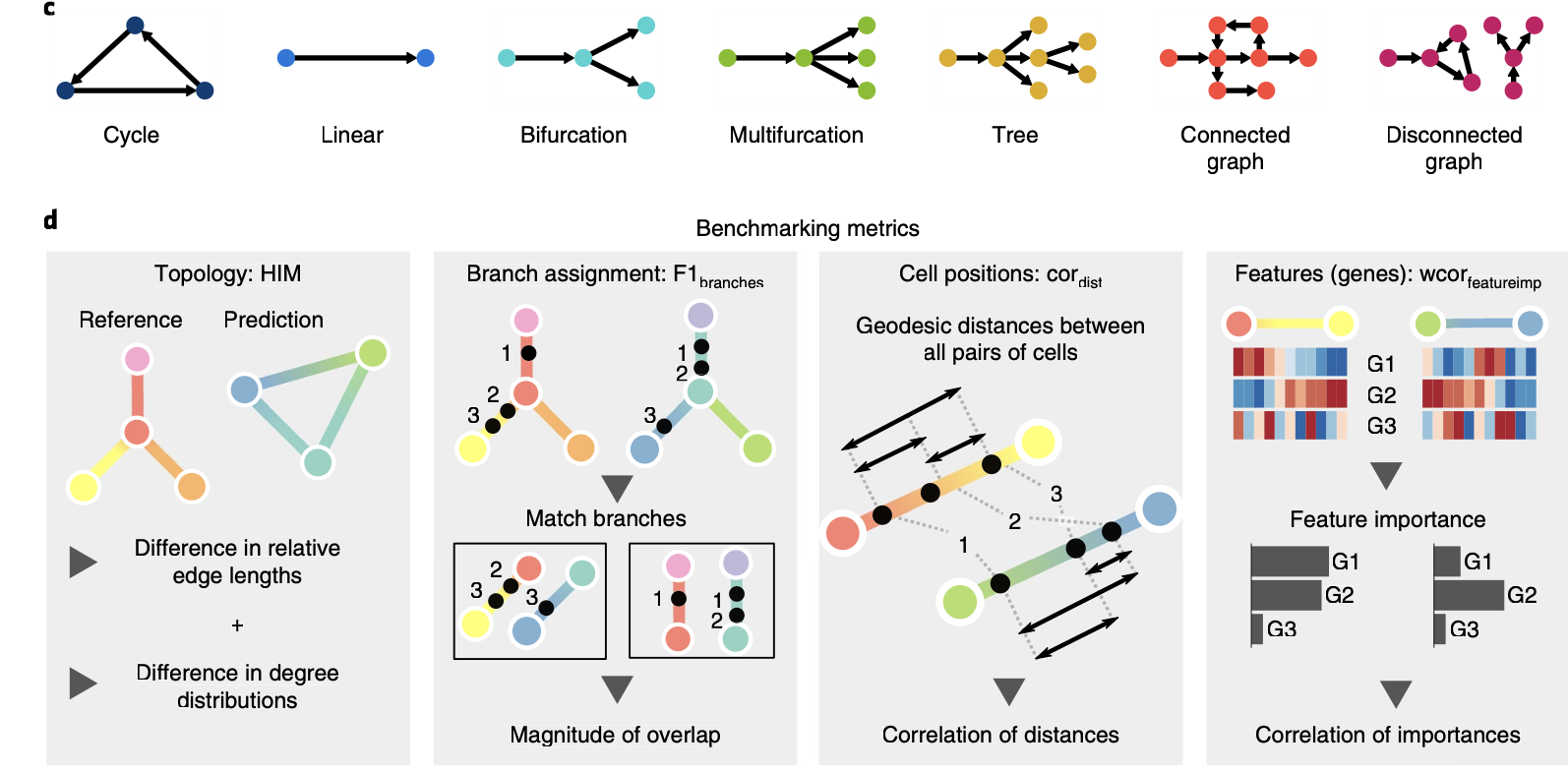
这个时候不同的拟时序软件工具就很难一次性覆盖这些全部的7种不同轨迹形式啦,不同的工具擅长不同的轨迹,这样的话对纯粹的工具使用者来说就很尴尬,因为大家不可能说在自己的单细胞项目去把这几十种工具全部测试一次,还得针对7种不同轨迹形式进行组合评价,工作量不可谓不大!
所以这个时候其实生物学背景更加重要了,前面我们在 为什么做拟时序 提到了其实可以把拟时序分析简化成为了主要是为了展示差异细节,比如CD14和CD16的两种单核细胞的差异细节,或者说说CD56和CD16这两种NK细胞的差异细节。我们的生物学背景让我们知道简单的两个细分亚群大概率上就是简单的Linear和branched形式的轨迹,不太可能是超级复杂的各种拟时序轨迹结果,除非你做的是完全陌生领域的陌生单细胞亚群的从零开始的真正的发育推断!
既然绝大部分小伙伴都做的是普普通通的差异细节的展示,那么其实根据这个综述选择一两个比较好的软件工具即可:
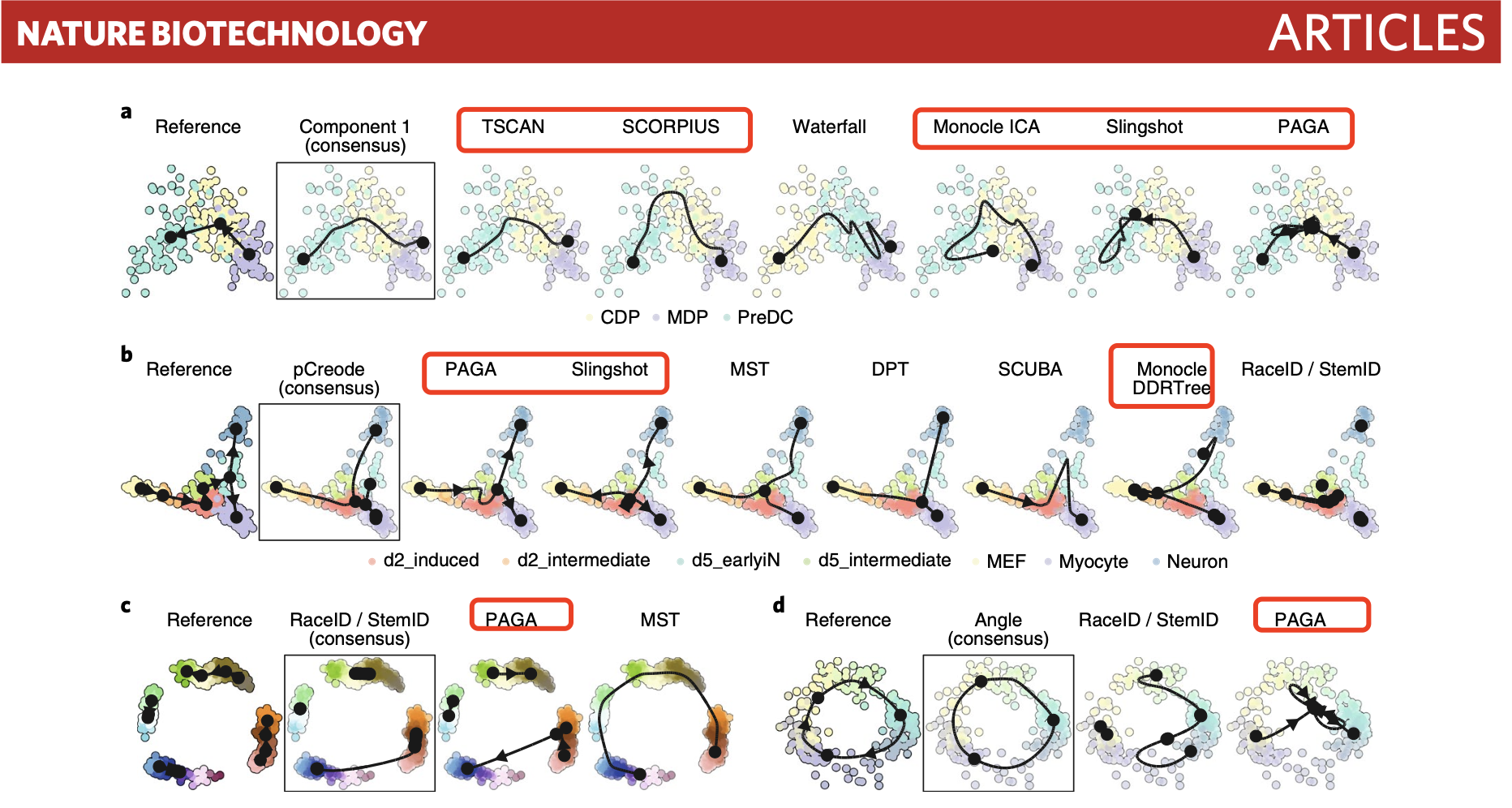
比如我们就分享了一下软件工具的实战笔记:
- 简单直接的拟时序分析方法,R包SCORPIUS推荐
- 把基因表达量画在拟时序结果图上
- 拟时序分析就是差异分析的细节剖析
- 拟时序分析的10个步骤
另外,值得一提的是这个2019的综述其实还把他们的45个软件的测评过程和结果做成了一个易于使用的R包dynverse is a collection of R packages aimed at supporting the trajectory inference (TI) :
- https://dynverse.org/
- https://github.com/dynverse/dynmethods
- https://github.com/dynverse/dyno
新的算法仍然会持续不断出来
可以看到早在2016的拟时序综述就已经确定下来了monocle2的江湖地位,到后来的2019综述的几十块拟时序软件就足以让普通用户眼花缭乱了,而且每个软件工具也会自我进化,比如monocle2的团队就更新了monocle3。
而且可以超脱表达量矩阵本身,引入全新信息来开发拟时序工具。比如可以引入具体的每个基因的转录本的未剪接和剪接 mRNA数值,就可以构建RNA 速率模型做分析,得到的发育顺序就有方向而且可靠啦,详见:10x官网下载pbmc3k数据集走RNA速率上下游分析实战。目前最经典的就是velocyto和scVelo啦,步骤是: - velocyto pipeline (La Manno et al., Nature, 2018) to obtain the pre-mature (unspliced) and mature (spliced) transcript information based on Cell Ranger output.
- 然后基于上面的loom文件去运行scVelo
参考:https://www.10xgenomics.com/cn/analysis-guides/trajectory-analysis-using-10x-Genomics-single-cell-gene-expression-data
但是这个基于基因的转录本的未剪接和剪接 mRNA数值的RNA速率推断也有缺点,首先是需要对测序的fastq文件重新分析,才能获取到未剪接和剪接 mRNA数值,其次在传统的基于二代测序数据的10x单细胞转录组其实仅仅是测了具体的每个mRNA的两端二选一而已,并没有把两个mRNA测通所以就很难获得准确的未剪接和剪接 mRNA数值。当然了,算法会一直有更新,全新的软件和工具也会被持续不断的开发出来,慢慢的就解决了以前的问题。比如2024新发表的scVelo,就不再依赖于剪接信息,而是通过引入基因调控数据 ,提供更准确的细胞轨迹信息和伪时间推断结果。详见 - 文章:TFvelo: gene regulation inspired RNA velocity estimation. Nat Commun
- github官网(Python软件):https://github.com/xiaoyeye/TFvelo
值得一提的是拟时序一本通专辑的目录是: - 为什么做拟时序 (展示差异细节)
- 拟时序的正确姿势,错误示范,创新型拟时序
- 拟时序的多种算法大比拼
- monocle2拟时序实战
- 降维聚类分群
- 拟时序
- 多种基础可视化
- 正向特殊可视化(目标基因表达量,基因集打分)
- 反方向特殊可视化(5种可视化)
- monocle3拟时序实战
- SCORPIUS实战
- Slingshot实战
- 基于其它编程语言的拟时序分析
然而,本人的理解肯定是片面的,希望通过这10个推文的指引能让大家对单细胞转录组的明星分析方法拟时序有一个基础的认知。也欢迎大家提出来自己的不同的看法,或者自己对拟时序的疑惑之处,欢迎留言参与讨论哦!