前面我们在 为什么做拟时序 提到了其实可以把拟时序分析简化成为了主要是为了展示差异细节,比如CD14和CD16的两种单核细胞的差异细节,或者说说CD56和CD16这两种NK细胞的差异细节。
也就是说,我们做拟时序之前通常是要细分亚群到足够深入,需要确定被做拟时序分析的对象是有比较大生物学变化的可能性。但是也有很多文章在使用拟时序分析的时候其实忽略这个前提,就显得很“可笑”,接下来我们就用3个案例来说明一下拟时序的正确姿势,错误示范,还有创新型的应用场景。
首先是正确的拟时序应用
可以看 2024发表在《Hepatology Communications.》杂志的单细胞转录组数据挖掘文章:《Single-cell landscape identifies the immunophenotypes and microenvironments of HBV-positive and HBV-negative liver cancer》,文章做完了第一层次降维聚类分群后,再次提取里面的T细胞进行细分亚群就有CD4,CD8等完全不一样的子集,然后是针对CD8这个t细胞亚群继续细分成为了两个亚群,如下所示的GZMH高表达的效应(毒性)T细胞和PDCD1高表达的耗竭T细胞,在(D) T-SNE 可以看到两个亚群是有很清晰的界限的,所以在(E). Dot plot 可以看到这两个亚群的差异基因的可视化情况。但是前面我们在 为什么做拟时序 提到了其实可以把拟时序分析简化成为了主要是为了展示差异细节,这个文章里面也是如此,就有了图(F)的monocle2的拟时序结果,因为我们关心的是GZMH高表达的效应(毒性)T细胞和PDCD1高表达的耗竭T细胞两者的变迁关系,所以作者依据monocle2的拟时序结果把两个细胞亚群的具体的细胞排序后就可以展示了cytotoxic genes or exhausted genes
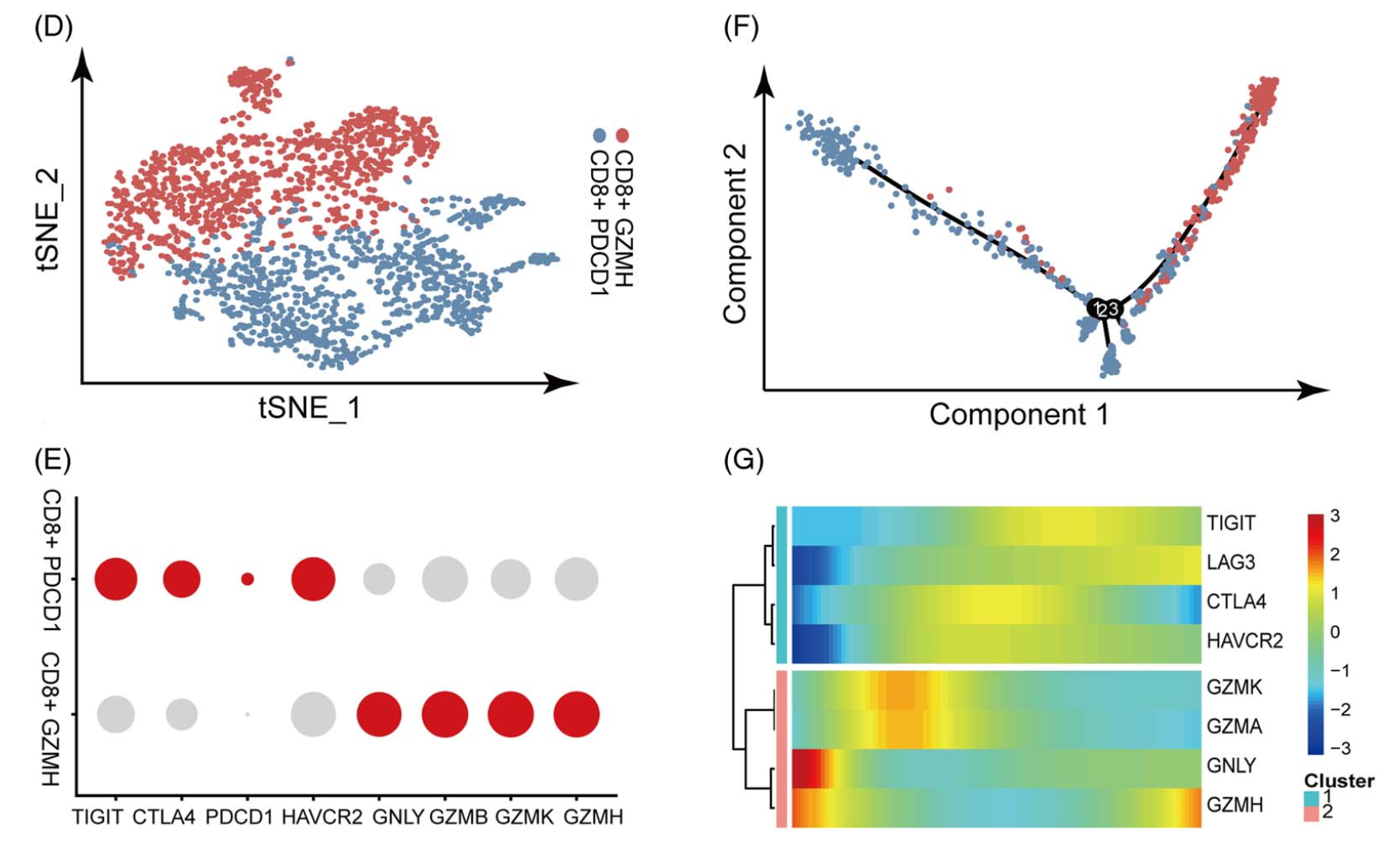
图例是:
- (D) T-SNE plot showing the annotation and color codes for two CD8+ T cell subclusters.
- (E). Dot plot showing the expression of canonically expressed genes in two subclusters.
- (F) Pseudotime-ordered analysis of CD8+ T cells from HBV+ HCC and HBV− samples. CD8+ T cell subtypes are labeled by colors.
- (G) Heat map showing the dynamic of cytotoxic genes or exhausted genes
非常清晰明了,这个完整的数据分析我们在 为什么做拟时序 演示了一个简化版,后面也会继续更新完整版!错误的拟时序分析例子
2024发表在《Journal of Cancer 》杂志的文章:《Integrated Bulk and Single-cell RNA Sequencing Data Constructs and Validates a Prognostic Model for Non-small Cell Lung Cancer》,文献进行了第一层次降维聚类分群后,并没有然后的生物学背景,就一股脑的把全部的肺癌肿瘤微环境里面的免疫细胞和间质细胞都拿去了拟时序,如下所示:
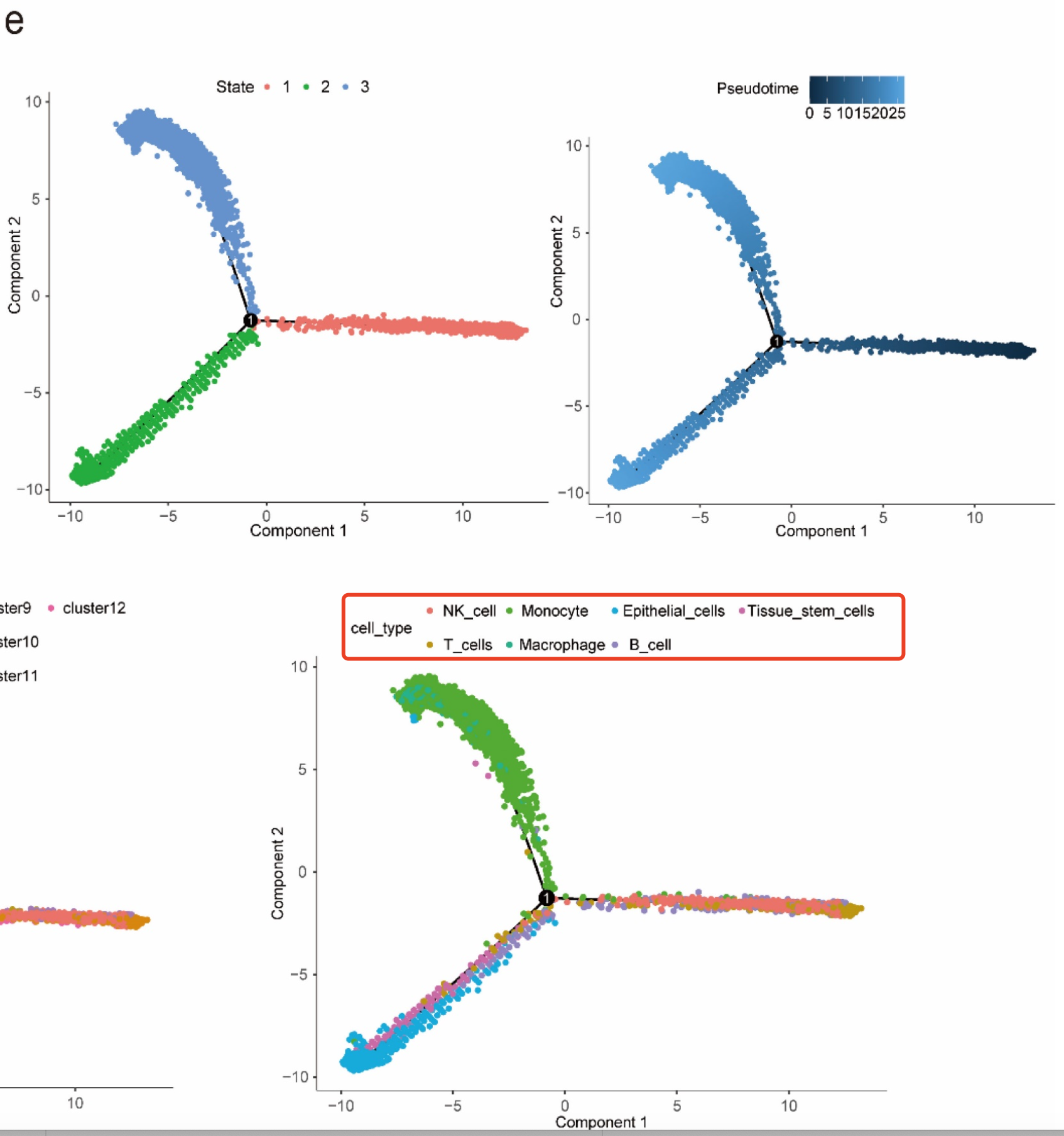
这样其实完全得不出什么结论,在肿瘤里面免疫细胞又不可能变迁成为恶性的上皮肿瘤细胞,就算是拿到了算法层面的每个细胞的拟时序分析后产生的 Pseudotime 和State,也很难去做生物学描述。一些拟时序创新
因为单细胞转录组数据的流行才让大家都认识到了这个拟时序分析可以展示差异分析的细节,但其实展现 差异分析细节这个需求在单细胞之前就存在。只不过呢,我们常规的bulk转录组通常是少量的有限的三五个组别,每个组就三五个样品,所以差异分析拿到了变化倍数和统计学指标就足够了,后面主要是针对差异基因集去做各种生物学功能富集去编造生物学故事啦。但是,TCGA或者GTEX等计划里面的常规的bulk转录组样品数量其实已经是成百上千了,我们的单细胞转录组的各个亚群其实里面也就是成百上千个细胞,两者从表达量矩阵格式来说并没有本质区别,都是可以走拟时序分析算法的。
比如2021的文章:《Preoperative immune landscape predisposes adverse outcomes in hepatocellular carcinoma patients with liver transplantation》就是一千多个样品,它们的两万多个基因构成了一个表达量矩阵,但是文章作者仅仅是关心那些5000个表达量比较高的蛋白质编码基因,然后就做了拟时序分析:
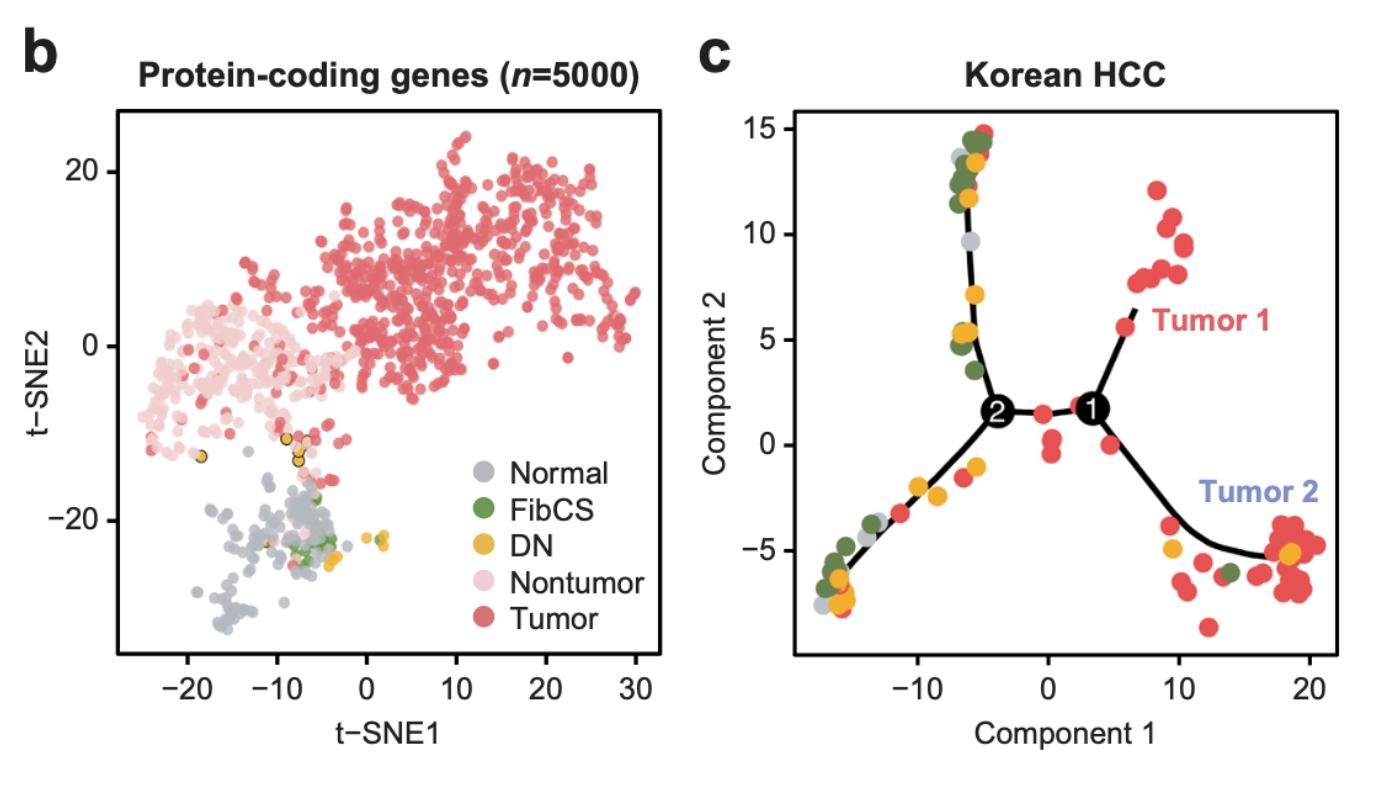
其实就是肿瘤样品可以区分成为两个分子分型,上面的图例是 : - b t-SNE analysis using the 5000 most highly expressed protein-coding genes in the meta-dataset (n = 1179).
- c Trajectory analysis of Korean HCC using Monocle 2.
而且可以很清晰的看到正常的乳腺组织跟两个分子分型的肿瘤样品的差异细节:
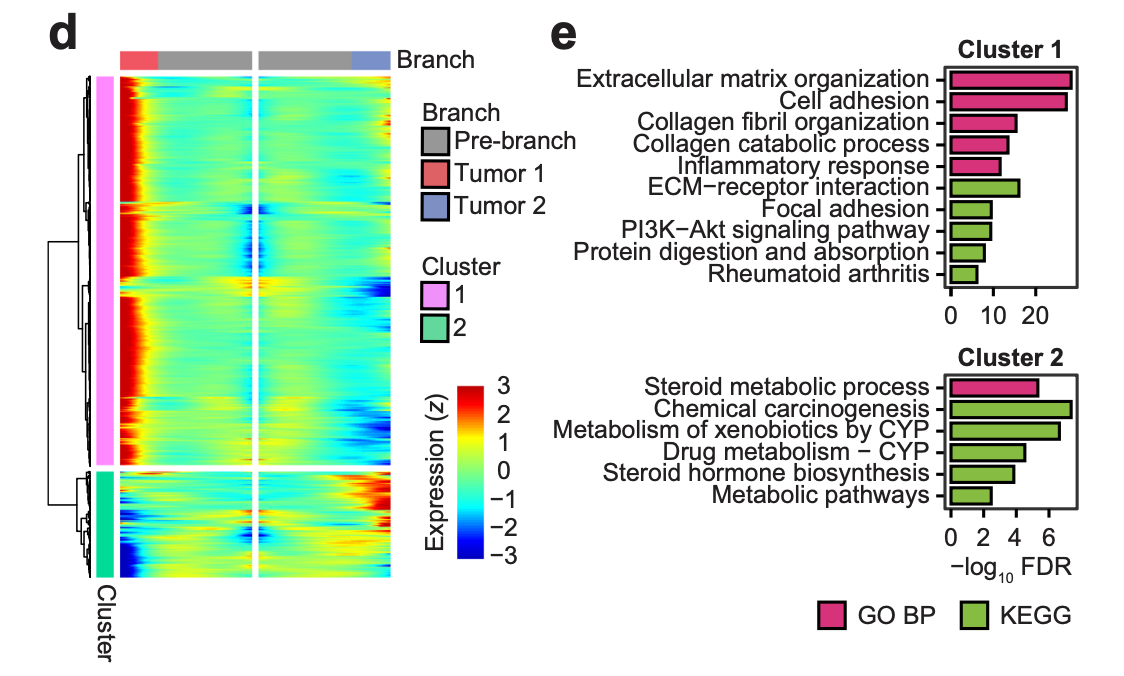
值得一提的是拟时序一本通专辑的目录是: - 为什么做拟时序 (展示差异细节)
- 拟时序的正确姿势,错误示范,创新型拟时序
- 拟时序的多种算法大比拼
- monocle2拟时序实战
- 降维聚类分群
- 拟时序
- 多种基础可视化
- 正向特殊可视化(目标基因表达量,基因集打分)
- 反方向特殊可视化(5种可视化)
- monocle3拟时序实战
- SCORPIUS实战
- Slingshot实战
- 基于其它编程语言的拟时序分析
然而,本人的理解肯定是片面的,希望通过这10个推文的指引能让大家对单细胞转录组的明星分析方法拟时序有一个基础的认知。也欢迎大家提出来自己的不同的看法,或者自己对拟时序的疑惑之处,欢迎留言参与讨论哦!