看到了一个在《Journal of Thoracic Oncology》期刊的研究,文章标题:《EGFR Oncogenic Mutations in NSCLC Impair Macrophage Phagocytosis and Mediate Innate Immune Evasion Through Upregulation of CD47》,研究者们将两种最常见的突变类型EGFR19del和EGFRL858R,分别转进三种不同的癌细胞系A549、H1299和Beas-2B(前两个是肺腺癌细胞系,第三个是正常的人肺上皮细胞系),发现引入突变型EGFR后,这些细胞系在蛋白和mRNA水平上都表现出CD47的显著上调。(一般来说,肿瘤细胞系都是纯纯的恶性的上皮细胞 )
而且还经过了一些肿瘤病人的转录组队列数据集同样的EGFR突变与否的分组后差异分析,也是有CD47作为多个数据集差异结果的交集,证据链非常solid:
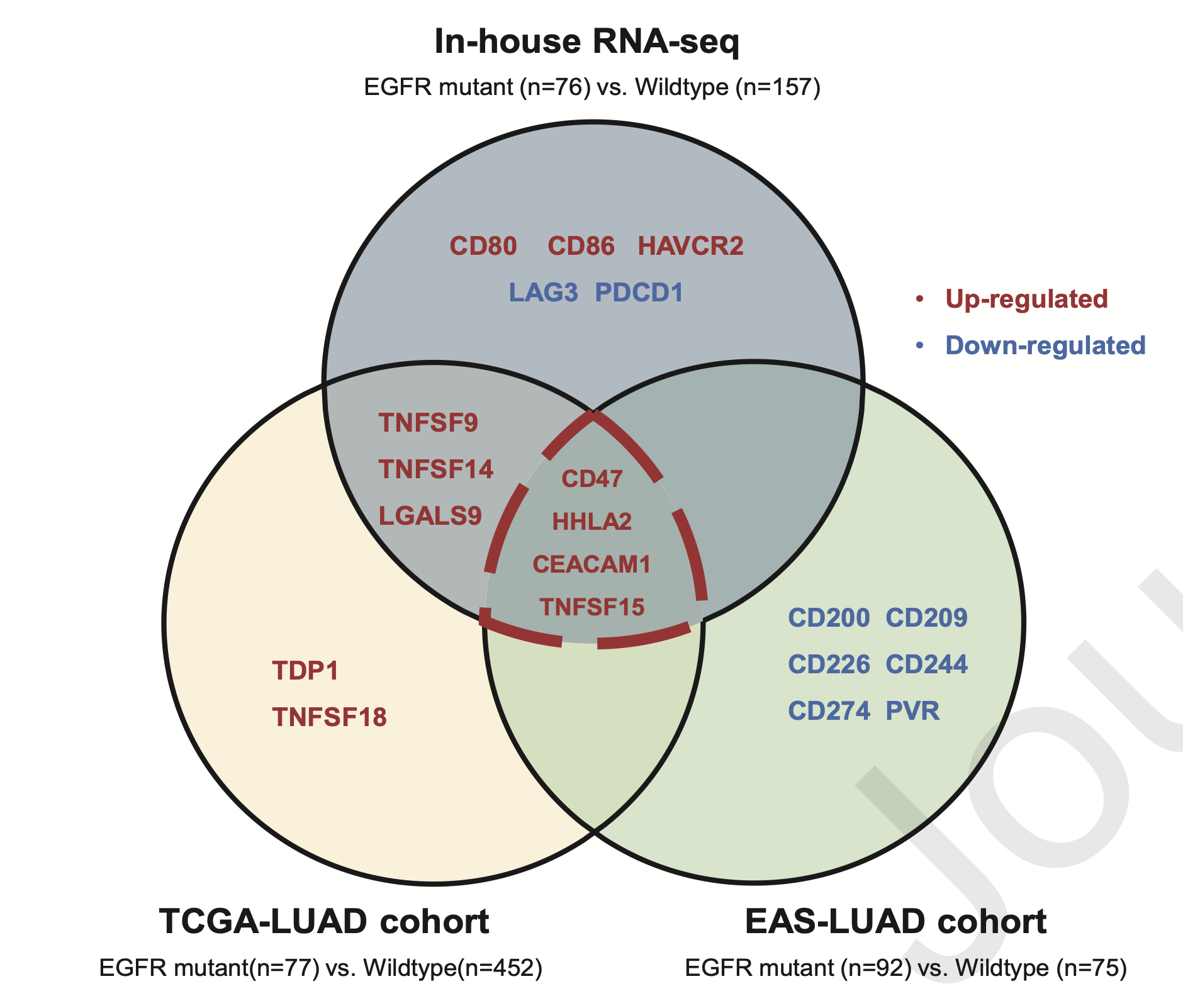
我看到了这个结论马上就想起来了,之前看到的数据挖掘文章:《Single-cell Analyses Reveal Tumor Microenvironment Differences between EGFR 19del and L858R mutations in Lung Adenocarcinoma》,作者从上面提到的GSE171145数据集里面拿到了 two 19del patients, two L858R patients and two wild-type patients ,我看了看前面GSE171145的文献里面的病人描述,确实有EGFR具体突变位点信息,文章的落脚点就是3个分组 ,分别是:
- 19del LUAD (10,688 cells),
- L858R LUAD (10,286 cells),
- EGFR-wild type LUAD (10,510 cells),
也就是说,这个是天然的病人分组,都没必要去做癌症细胞系实验啦,只需要看看CD47这个基因是否在EGFR突变肺腺癌病人组比EGFR野生型肺腺癌病人组高表达即可,而且还可以看看如果CD47这个基因确实是在EGFR突变肺腺癌病人组高表达的话,它是在具体的哪个单细胞亚群高表达呢?GSE171145这个数据集的表达量矩阵如下所示:
ls -lh *counts*|cut -d" " -f7-
3.5M 3 26 2021 GSM5219674_LJQ-T.counts.tsv.gz
14M 3 26 2021 GSM5219675_GBG-T.counts.tsv.gz
14M 3 26 2021 GSM5219676_LYB-T1.counts.tsv.gz
12M 3 26 2021 GSM5219677_LYB-T2.counts.tsv.gz
9.9M 3 26 2021 GSM5219678_CYD-T.counts.tsv.gz
12M 3 26 2021 GSM5219679_CYZ-T.counts.tsv.gz
12M 3 26 2021 GSM5219680_XMS.counts.tsv.gz
9.1M 3 26 2021 GSM5219681_ZYQ.counts.tsv.gz
10M 3 26 2021 GSM5219682_TGS.counts.tsv.gz
很容易批量读取这些不同样品表达量矩阵文件:
library(data.table)
dir='GSE171145_RAW/'
samples=list.files( dir ,pattern = '.counts.tsv.gz')
samples
library(data.table)
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
ct=fread(file.path( dir ,pro),data.table = F)
ct[1:4,1:4]
rownames(ct)=ct[,1]
ct=ct[,-1]
sce=CreateSeuratObject(counts = ct ,
min.cells = 5,
min.features = 300 )
return(sce)
})
do.call(rbind,lapply(sceList, dim))
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples )
names(sce.all@assays$RNA@layers)
sce.all[["RNA"]]$counts
# Alternate accessor function with the same result
LayerData(sce.all, assay = "RNA", layer = "counts")
sce.all <- JoinLayers(sce.all)
dim(sce.all[["RNA"]]$counts )
然后走我们的标准代码,常规的单细胞转录组降维聚类分群代码可以看 链接: https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh ,基本上大家只需要读入表达量矩阵文件到r里面就可以使用Seurat包做全部的流程,但是初始情况下只能说是拿到如下所示的降维聚类分群图:
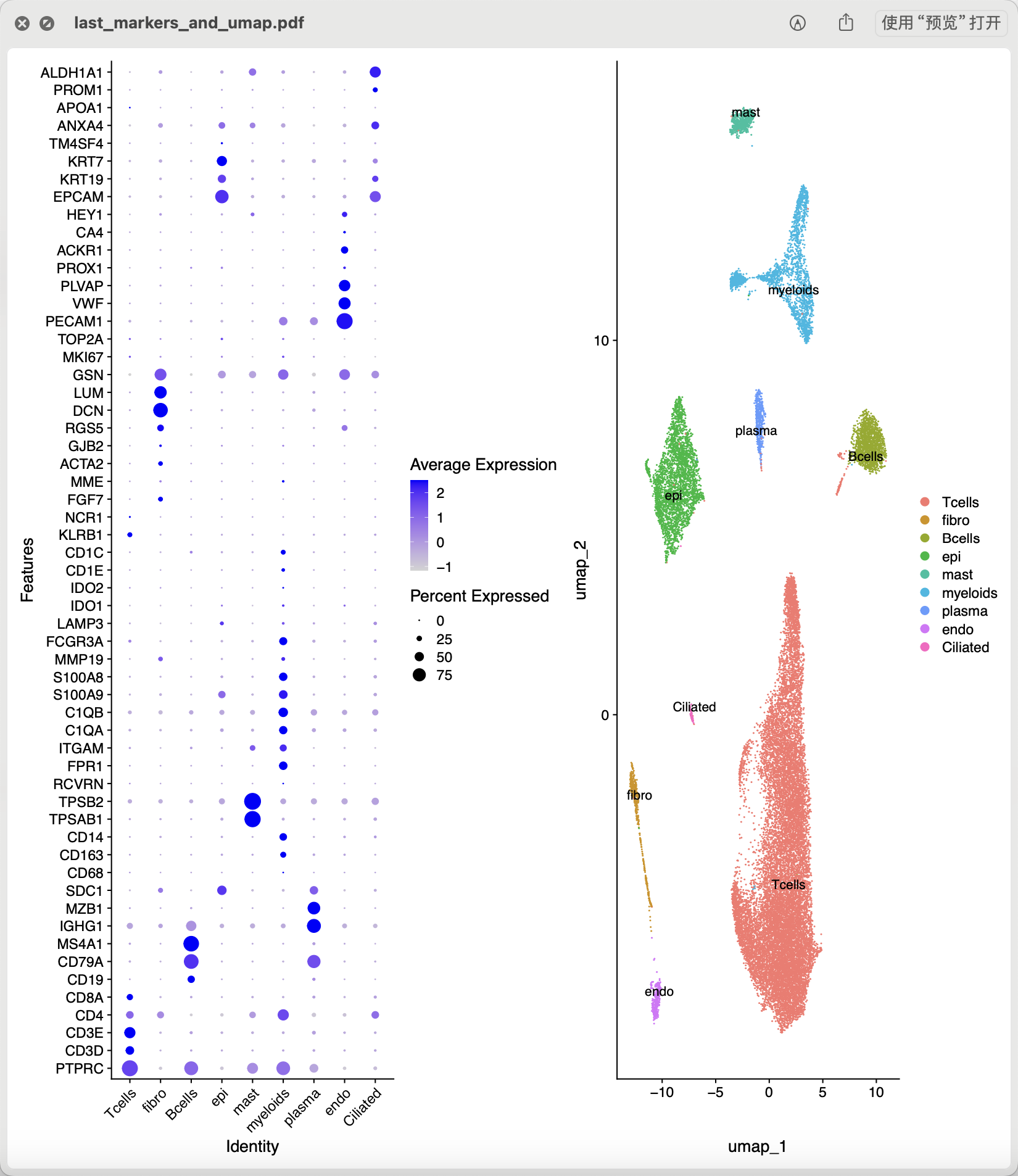
值得注意的是GSE171145这个数据集的作者并没有说清楚具体的每个单细胞表达量矩阵来源于的病人的突变情况,在附件也看不到,但是我们仍然是可以检查一下CD47这个基因的表达量,很简单的小提琴图即可;
sce.all = readRDS('./2-harmony/sce.all_int.rds')
sp='human'
load('./phe.Rdata')
rownames(phe) = colnames(sce.all)
sce.all@meta.data = phe
sel.clust = "celltype"
sce.all <- SetIdent(sce.all, value = sel.clust)
table(sce.all@active.ident)
DimPlot(sce.all)
colnames(sce.all@meta.data)
table(sce.all$celltype)
VlnPlot(sce.all[,sce.all$celltype=='epi'],
'CD47',pt.size = 0,split.by = 'orig.ident') + ggsci::scale_fill_igv()
可以看到,确实是这8个病人里面的上皮细胞里面的CD47基因表达量是区别的,但是因为GSE171145这个数据集的作者没有说清楚分组,就有点麻烦了 :
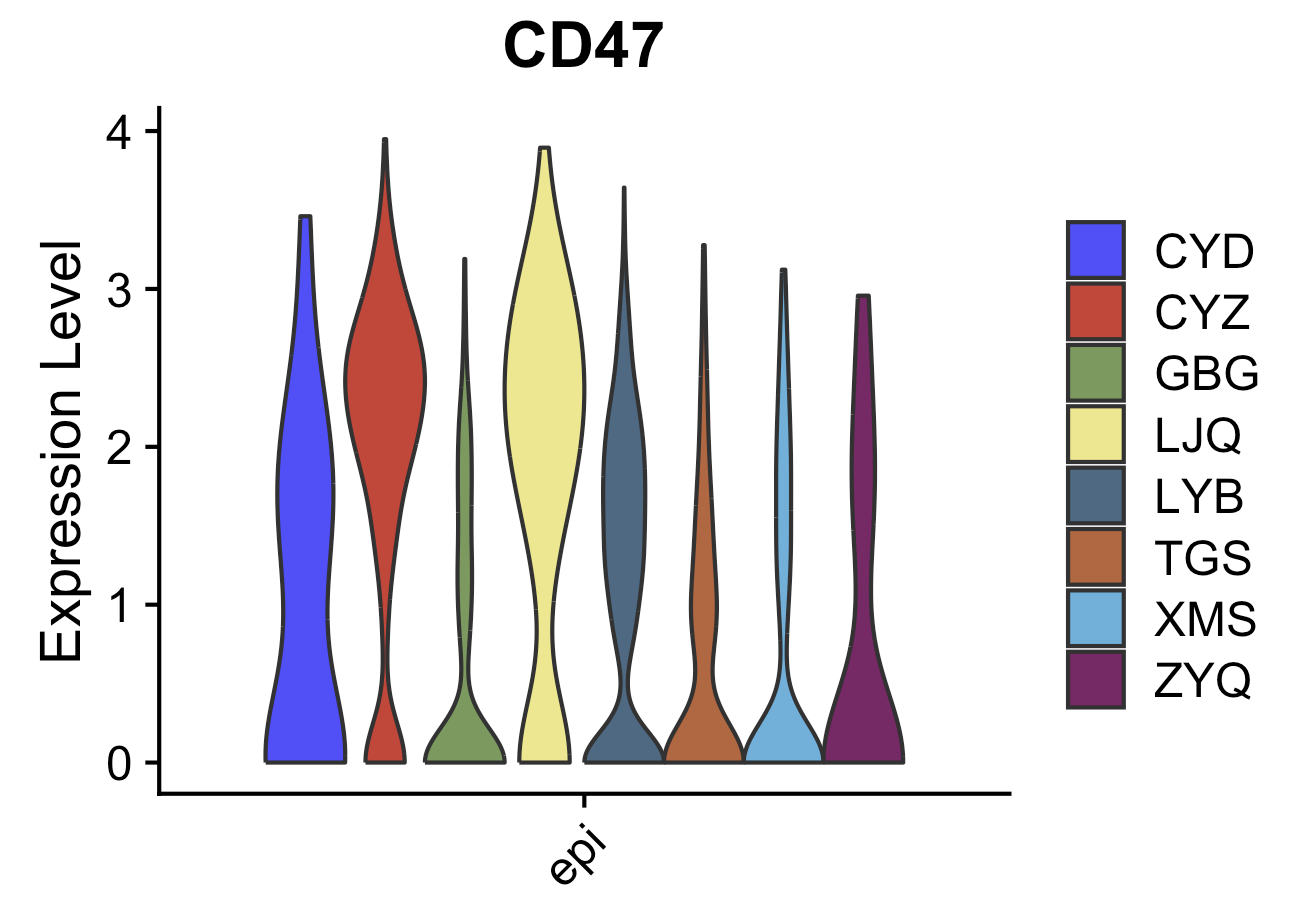
我发了邮件给GSE171145这个数据集的作者,但是也没有人回复我:
Contact name Qing Zhou E-mail(s) gzzhouqing@126.com
Organization name Guangdong Lung Cancer Institute, Guangdong Provincial People's Hospital, Guangdong Academy of Medical Sciences, Guangzhou, China
Street address 106#,ZHONG SHAN ER ROAD
City Guangzhou
State/province Guangdong
ZIP/Postal code 510010
Country China
学徒作业
肺癌的单细胞数据集那么多,可以做一个系统性梳理,看看是否还有其它公共数据集也同时提供了病人的EGFR突变信息,然后走同样的降维聚类分群后,针对里面的恶性上皮细胞看看CD47基因表达量,看看是否能得到同样的结果!
提供服务器完成学徒作业哈,只需要你承诺是完成我们的学徒作业专辑里面的任意习题,都可以获得服务器账号一个,详情欢迎进交流群了解哈!
关于生信游民
学徒作业专辑其实对应的是我一直提倡的生信游民理念,咱们做数据处理的小伙伴理论上是可以在全球任意地方远程办公,只要你的技术够硬责任心足够好,在接单市场上应该是所向披靡啦。前提是你如何展现自己,我们的生信游民系列练习题就是一个很好的渠道,你只需要任意完成其中几个就能对外show出你的数据处理能力了,群里就有数不胜数的兼职项目等你来拿 :
- 凭感觉这个数据挖掘文章里面的差异基因上下调应该是弄反了
- 处理前后单细胞转录组数据的整合与否确实影响了分析结论
- 凭什么说TCGA和GEO数据挖掘的结论不可靠呢
- 转录组差异分析不足以说明你的目标基因调控某个通路
- 五年高考三年模拟的R包
- 史上最大子宫内膜异位症单细胞图谱
- 只有测序数据不知道如何高级分析可以试试看这个期刊