我在生信技能树的早期教程:《你确定你的差异基因找对了吗?》提到过,必须要对你的转录水平的全局表达矩阵做好质量控制,最好是看到标准3张图:
- 左边的热图,说明我们实验的两个分组,normal和npc的很多基因表达量是有明显差异的
- 中间的PCA图,说明我们的normal和npc两个分组非常明显的差异
- 右边的层次聚类也是如此,说明我们的normal和npc两个分组非常明显的差异
如果分组在3张图里面体现不出来,实际上后续差异分析是有风险的。这个时候需要根据你自己不合格的3张图,仔细探索哪些样本是离群点,自行查询中间过程可能的问题所在,或者检查是否有其它混杂因素,都是会影响我们的差异分析结果的生物学解释。有了合理的分组才可以进行简单的差异分析流程,基本上转录组测序技术和芯片技术拿到的表达量矩阵后续分析大同小异,公众号推文在:
但是因为数据挖掘的核心是缩小目标基因,所以大家越来越喜欢强行找差异。各种数据挖掘文章本质上都是要把目标基因集缩小,比如表达量矩阵通常是2万多个蛋白编码基因,不管是表达芯片还是RNA-seq测序的,采用何种程度的差异分析,最后都还有成百上千个目标基因。如果是临床队列,通常是会跟生存分析进行交集,或者多个数据集差异结果的交集,比如:多个数据集整合神器-RobustRankAggreg包 ,这样的基因集就是100个以内的数量了,但是仍然有缩小的空间,比如lasso等统计学算法,最后搞成10个左右的基因组成signature即可顺利发表。其实还有另外一个策略,有点类似于人工选择啦,通常是可以往热点靠,比如肿瘤免疫,相当于你不需要全部的两万多个基因的表达量矩阵进行后续分析,仅仅是拿着几千个免疫相关基因的表达矩阵即可。
现在比较流行的Olink蛋白质组技术,就恰好符合这两个策略,首先它是人工选择了几百个或者几千个有明确功能的蛋白质进行检测,其次它在很多研究里面其实是在强行找差异,比如2024年3月的文章:《Serum Olink Proteomics-Based Identification of Protein Biomarkers Associated with the Immune Response in Ischemic Stroke》,超级简单的一个实验设计 :
- Here, we used Olink proteomics to examine the expression levels of 92 immune response-related proteins in the sera of IS patients (n = 88) and controls (n = 88),
- we found that 59 of these proteins were differentially expressed.
有了差异列表就可以通过一系列机器学习,比如文章里面的least absolute shrinkage and selection operator (LASSO) and the random forest 去挑选最重要的元素,这个文章挑出来的是 MASP1, STC1, HCLS1, CLEC4D, PTH1R, and PIK3AP1, 可以比较好的区分疾病组和对照组!
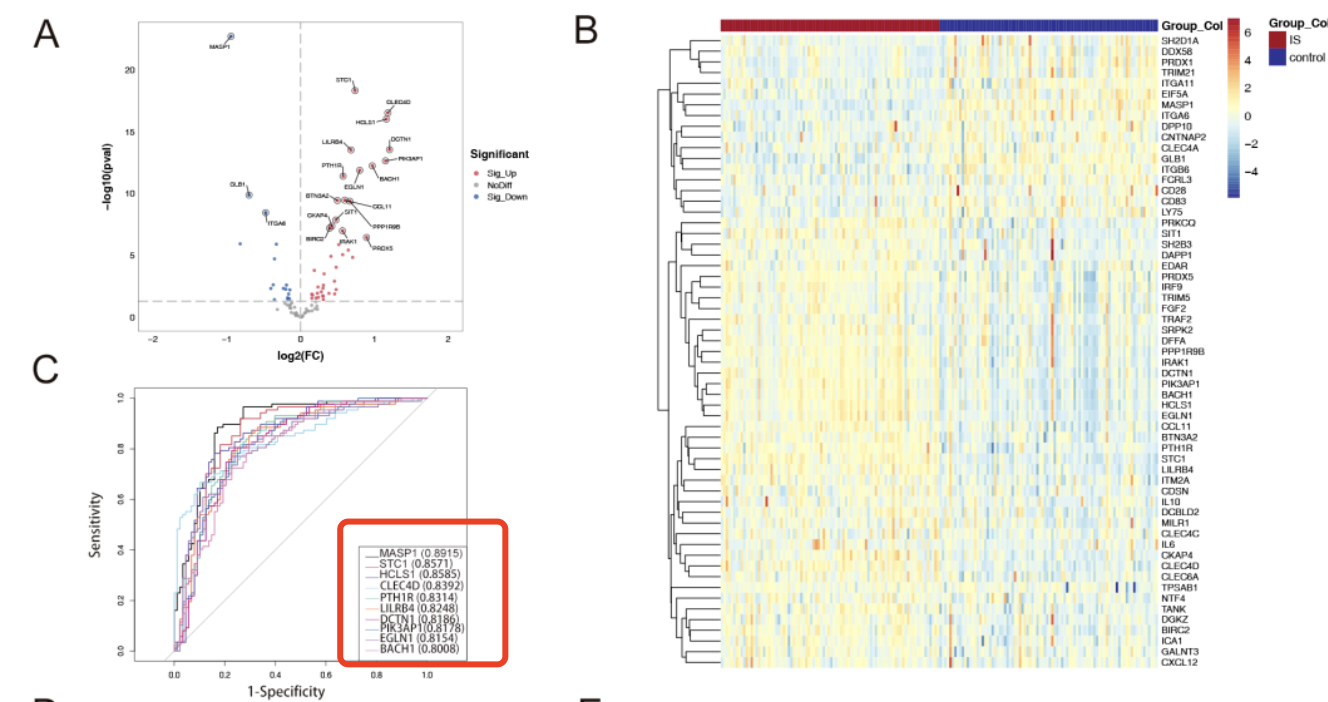
生信技能树的早期教程:《你确定你的差异基因找对了吗?》提到过,如果是表达量矩阵,就必须要有三张图,但是这个文章就没有。不过这个研究仅仅是检测92 immune response-related proteins 就可以发现里面的 42个蛋白在IS患者中显著上调,而17个蛋白显著下调,所以原则上两个IS patients (n = 88) and controls (n = 88), 的组间差异应该是很大的。但是文章并没有给出来表达量矩阵,在文献附件也找不到 :
- Specific information on 92 immune response-related proteins in the discovery set (Table S1);
- GO terms enrichment of DEPs (Table S2);
- KEGG pathways enrichment of DEPs (Table S3); PPI analysis of DEPs (Table S4);
- collinearity tests of 6 proteins (Table S5); and logistic regression model in different sets of performance indicators (Table S6)
而且,本来就是设计的靶向蛋白质组看的就是免疫相关的不到100个蛋白质,从里面挑选一半的蛋白质去进行生物学功能数据库注释,难道结果不应该是已知的吗,从这92 immune response-related proteins 随机抽样100次,1000次,每次抽50个然后去注释,不都是没有相关的功能吗?
然后,我搜索到了另外一个类似的实验设计,不是Olink这样的靶向蛋白质组,是 SWATH-MS 这个蛋白质组技术,然后队列是 20 healthy controls and 20 ischemic stroke patients
- Using the information-dependent acquisition (IDA) mode, proteins in the samples were identified. Search parameters were as follows: TOF/MS survey was performed in the mass range of 250–2000 m/z.
- LC-MS/MS raw data were processed using the ProteinPilot v5.0 search engine (AB SCIEX) for the identification of peptides and proteins (Release April 2016).
如下所示的差异分析和富集分析 :
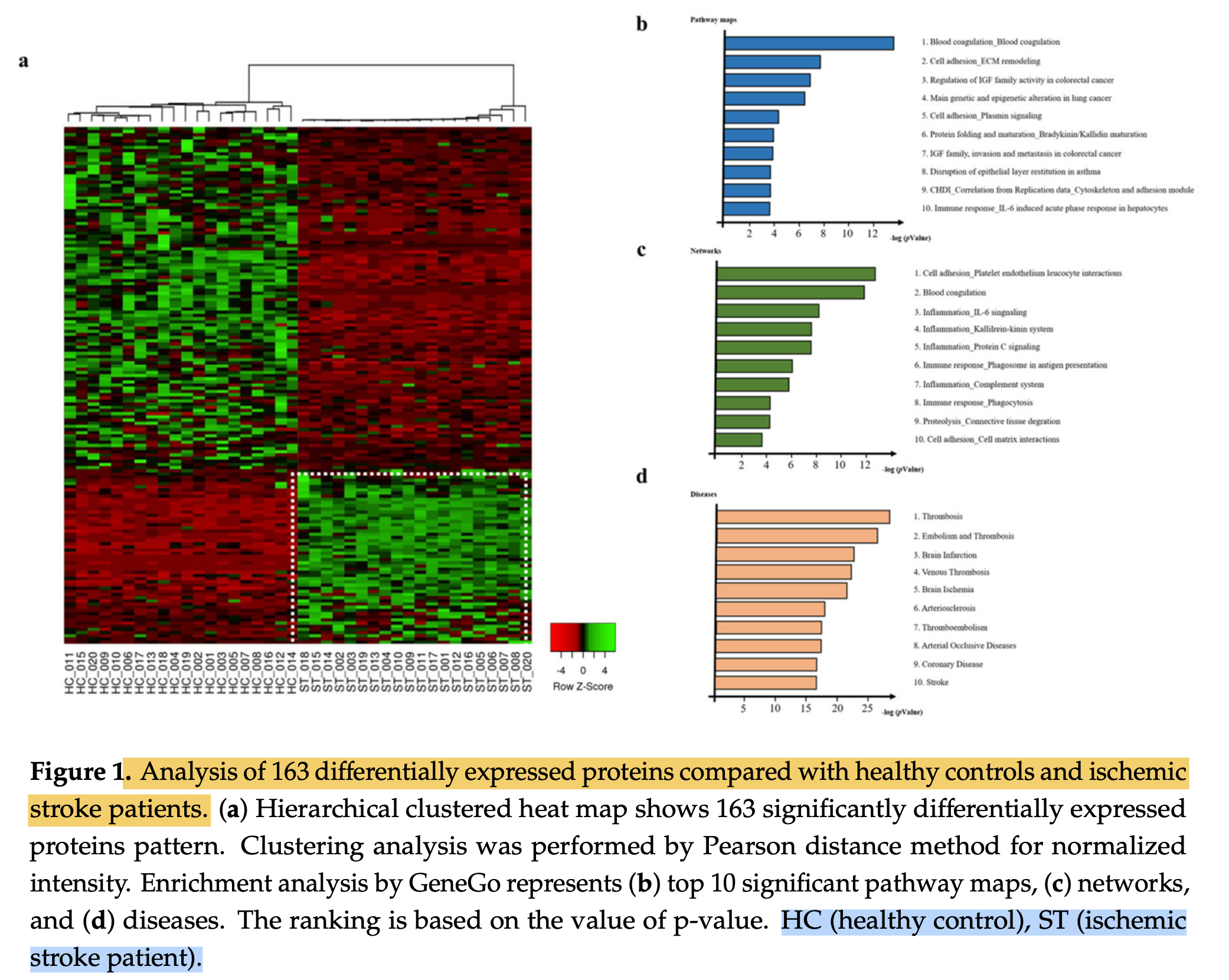
从这些差异的蛋白质里面定位到了13个目标:
Prothrombin
Coagulation factor IX Plasminogen
Fibrinogen alpha chain Fibronectin
Vitronectin
Histidine-rich
glycoprotein
Vitamin K-dependent protein S
Thrombospondin-1
Complement C1s subcomponent
Glutathione peroxidase 3
让我们熟悉一下这个SWATH-MS(Sequential Window Acquisition of All Theoretical Mass Spectra)技术吧,它是一种质谱技术,用于定量分析复杂样品中的蛋白质。SWATH-MS结合了两种质谱技术:数据依赖性采集(Data-Dependent Acquisition,DDA)和数据独立性采集(Data-Independent Acquisition,DIA),以实现高通量的蛋白质定量分析。SWATH-MS 的工作流程通常包括以下几个步骤:
- 样品制备: 样品制备是任何质谱实验的第一步。通常涉及蛋白质提取、消化(例如胰蛋白酶消化)和标记(如TMT或iTRAQ)等步骤。
- 质谱数据采集: SWATH-MS 首先进行数据依赖性采集(DDA),以确定样品中存在的肽段和蛋白质。然后,建立一个肽段库,记录每个肽段的质谱特征(如保留时间、质荷比等)。
- SWATH 扫描设置: 在 SWATH 扫描设置中,整个质谱质量范围被分成多个窗口。每个窗口的大小和数量取决于实验条件和仪器性能。
- 数据独立性采集(DIA): 在 SWATH-MS 实验中,每个窗口的数据独立性采集(DIA)扫描顺序。在每个窗口中,质谱仪会扫描整个质谱质量范围,并记录每个肽段的碎片谱。这样可以获得一个完整的肽段谱库。
- 数据分析: SWATH-MS 数据的分析通常包括两个主要步骤:(1)肽段和蛋白质的识别与定量,通常使用肽段库来与实验数据匹配,并量化每个肽段的丰度;(2)统计分析和生物信息学分析,用于识别不同条件之间的差异表达蛋白质,并分析蛋白质在生物过程中的功能和调节。
学徒作业
我相信这个 Ischemic Stroke 肯定是有海量的多组学数据,甚至单细胞多组学数据,理论上都是疾病组和对照组做一个简单的差异分析即可,就会有上下调基因就可以构建诊断模型。
大家首先做一个 Ischemic Stroke 的文献整理,梳理出来数据集列表,然后挑选里面的有表达量矩阵的文件进行上面的文章的同样的处理《Serum Olink Proteomics-Based Identification of Protein Biomarkers Associated with the Immune Response in Ischemic Stroke》,看看是否有交集吧!