前面我们演示了:10x技术空间单细胞上游定量案例分享(E-MTAB-12043),但是spaceranger定量失败。幸运的是很多懂行的小伙伴留言指出来了问题所在, 就是缺一个ffpe空间切片的探针文件。探针文件对于spaceranger的定量过程至关重要,因为spaceranger需要知道哪些探针与哪些转录本相关联,以便对空间位置上的RNA进行定量。
可以看到,官网上面说了它是 Input for spaceranger count pipeline
还是同样的代码即可,每个样品对应的fastq文件如下所示:
$ ls -lh *gz|cut -d" " -f 5-
1.1G 3月 4 21:33 Sample1_S1_L001_R1_001.fastq.gz
1.6G 3月 4 01:21 Sample1_S1_L001_R2_001.fastq.gz
1.2G 3月 4 04:02 Sample1_S2_L001_R1_001.fastq.gz
1.7G 3月 4 04:07 Sample1_S2_L001_R2_001.fastq.gz
1.2G 3月 4 02:48 Sample1_S3_L001_R1_001.fastq.gz
1.8G 3月 4 02:53 Sample1_S3_L001_R2_001.fastq.gz
1.1G 3月 4 02:56 Sample1_S4_L001_R1_001.fastq.gz
1.7G 3月 4 03:01 Sample1_S4_L001_R2_001.fastq.gz
写一个脚本文本文件( run_sapceranger.sh ),如下所示:
#! /bin/bash -xe
##
bin=/home/jmzeng/x10/pipeline/spaceranger-2.1.1/spaceranger
db=/home/jmzeng/x10/pipeline/refdata-gex-GRCh38-2020-A
ls $bin $db
fq_dir=${2}
/usr/bin/time -v $bin count --id=${1} \
--transcriptome=$db \
--fastqs=$fq_dir \
--probe-set=./Visium_Human_Transcriptome_Probe_Set_v1.0_GRCh38-2020-A.csv \
--sample=${1} \
--image=${3} \
--unknown-slide visium-1 \
--localcores=2 \
--localmem=80
上面的脚本文本文件( run_sapceranger.sh ),可以接受3个参数,包括样品的fastq格式的测序数据文件前缀,样品的fastq格式的测序数据文件所在的文件夹路径,以及样品的图片。我们来运行第一个样品,就是单个样品跑上面的脚本文本文件( run_sapceranger.sh )的代码:
nohup bash run_sapceranger.sh Sample1 \
./rawdata \
./Sample1.tif \
1>log_Sample1.txt 2>&1 &
我们是多个样品有规则的文件,所以可以写一个批处理:
for i in {1..7}; do
nohup bash run_sapceranger.sh "Sample${i}" ./rawdata "./Sample${i}.tif" 1> "log_Sample${i}.txt" 2>&1 &
done
当然了,这一切代码成功运行的前提是,文件是齐全并且完整的,空间单细胞转录组需要图片和fastq文件哦!这些文件都很大, 所以很容易下载失败,详见:aspera的高速下载确实很快吗。
这次是成功了,依然是根据前面演示的代码进行文件夹整理:10x技术空间单细胞上游定量案例分享(E-MTAB-12043),如下所示我们成功的案例是:
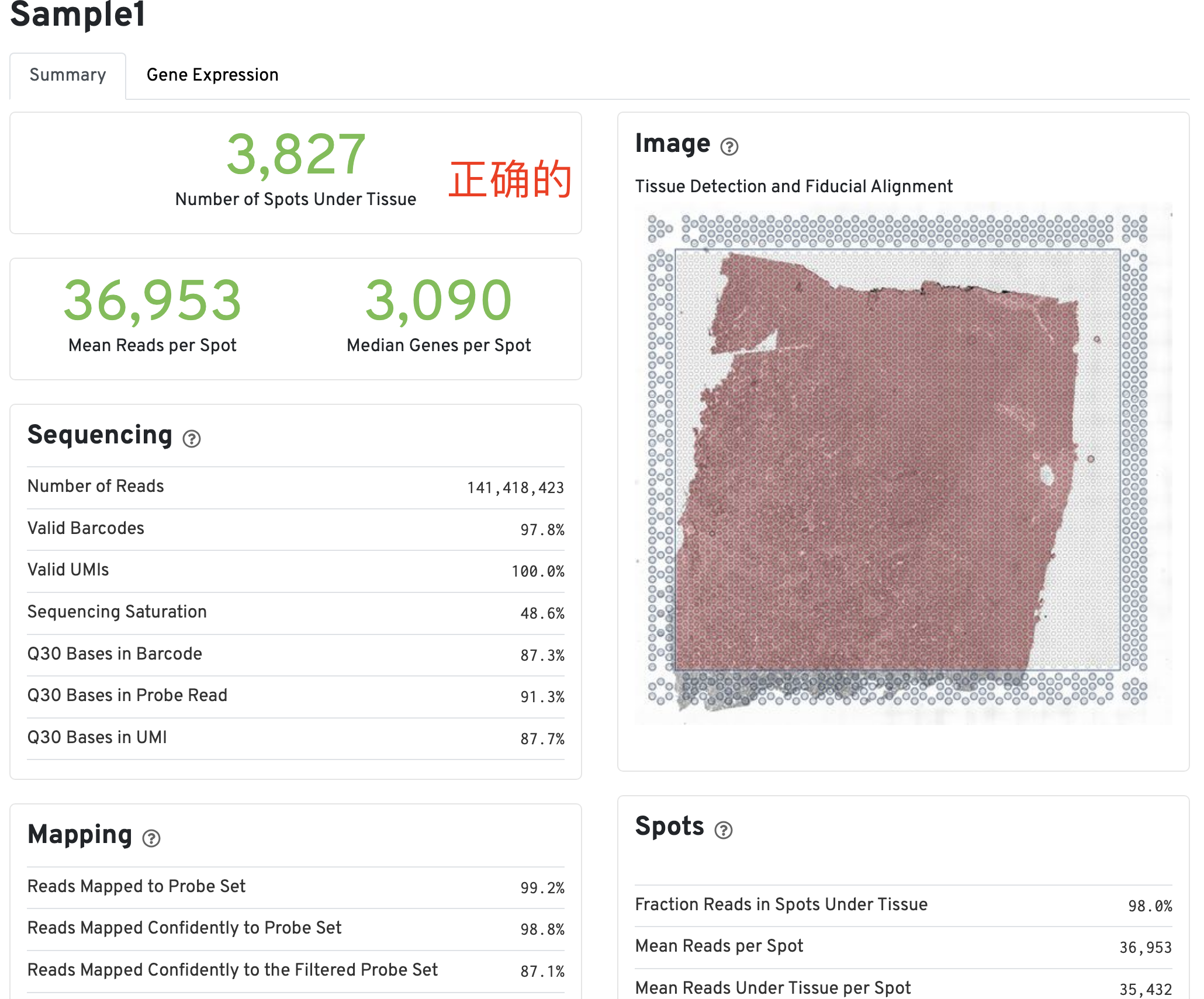
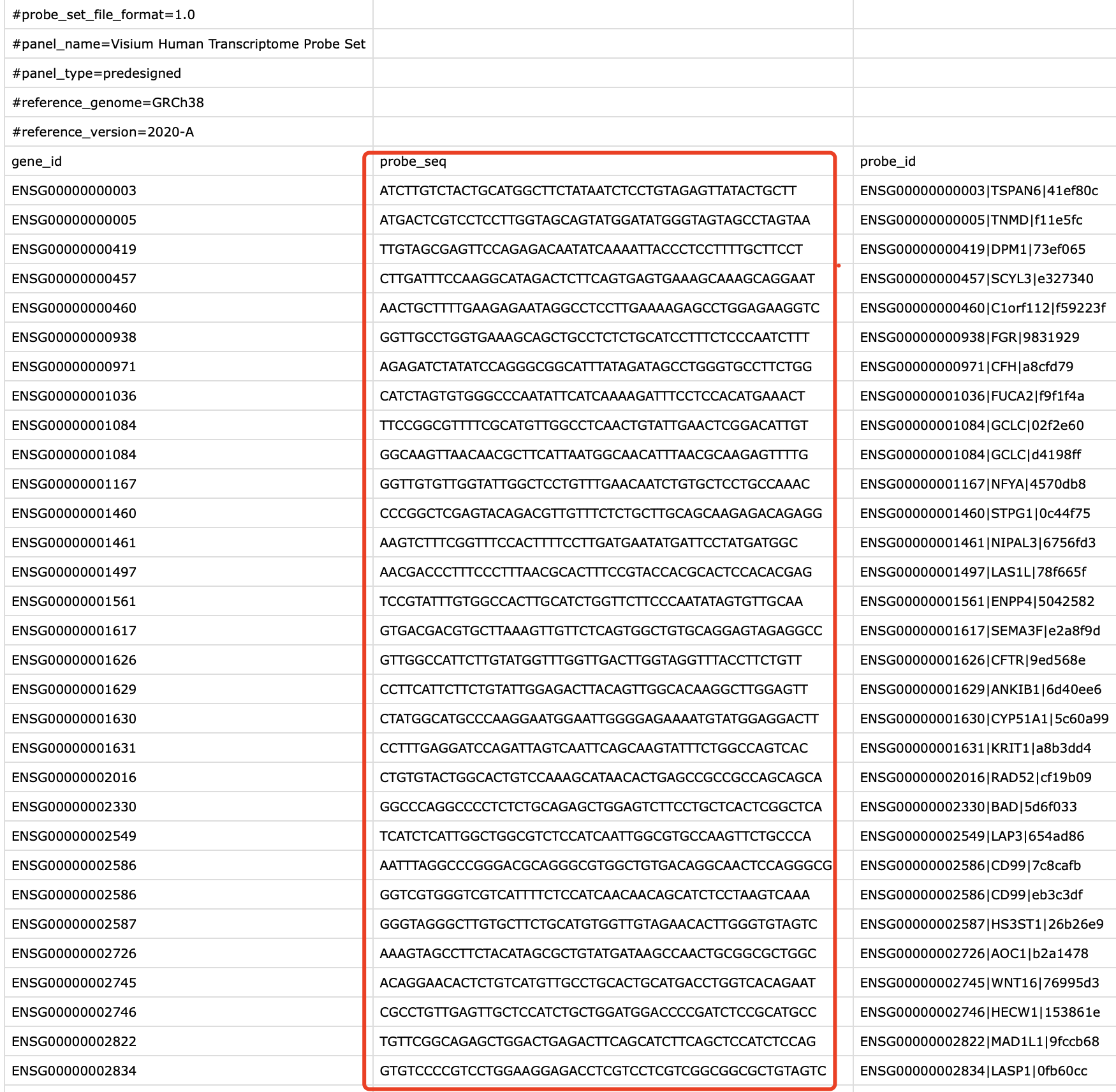
也就是说,仅仅是引入了一个 探针的csv文件,定量代码就ok了,这个探针文件如下所示;
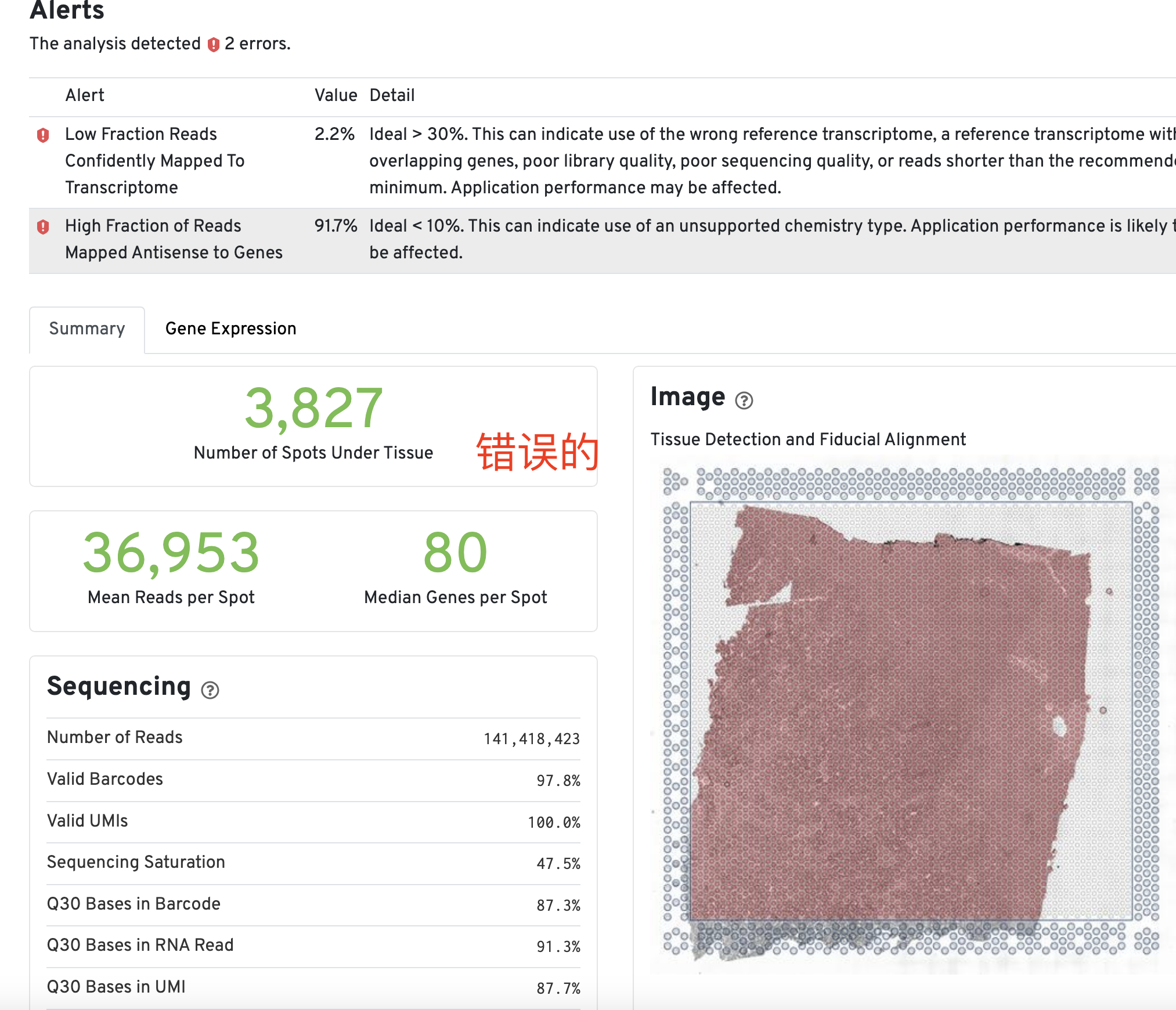
如果没有指定这个探针文件,就会失败了,如下所示是一个失败的案例:
值得一提的是,之前我们复现另外一个文章的时候,也是从fastq文件开始, 走spaceranger定量流程,详情请见:空间单细胞的上游定量流程(spaceranger,10x技术),它就不需要同样的探针csv文件啦。