通常我们拿到了肿瘤相关的单细胞转录组的表达量矩阵后的第一层次降维聚类分群通常是:
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibro or CD31+,PECAM1,endo)
参考我前面介绍过 CNS图表复现08—肿瘤单细胞数据第一次分群通用规则,这3大单细胞亚群构成了肿瘤免疫微环境的复杂。绝大部分文章都是抓住免疫细胞亚群进行细分,包括淋巴系(T,B,NK细胞)和髓系(单核,树突,巨噬,粒细胞)的两大类作为第二次细分亚群。但是也有不少文章是抓住stromal 里面的 fibro 和endo进行细分,并且编造生物学故事的。如下所示:

其实更麻烦的地方在于,我们第一层次降维聚类分群的时候往往是比较容易复现的:
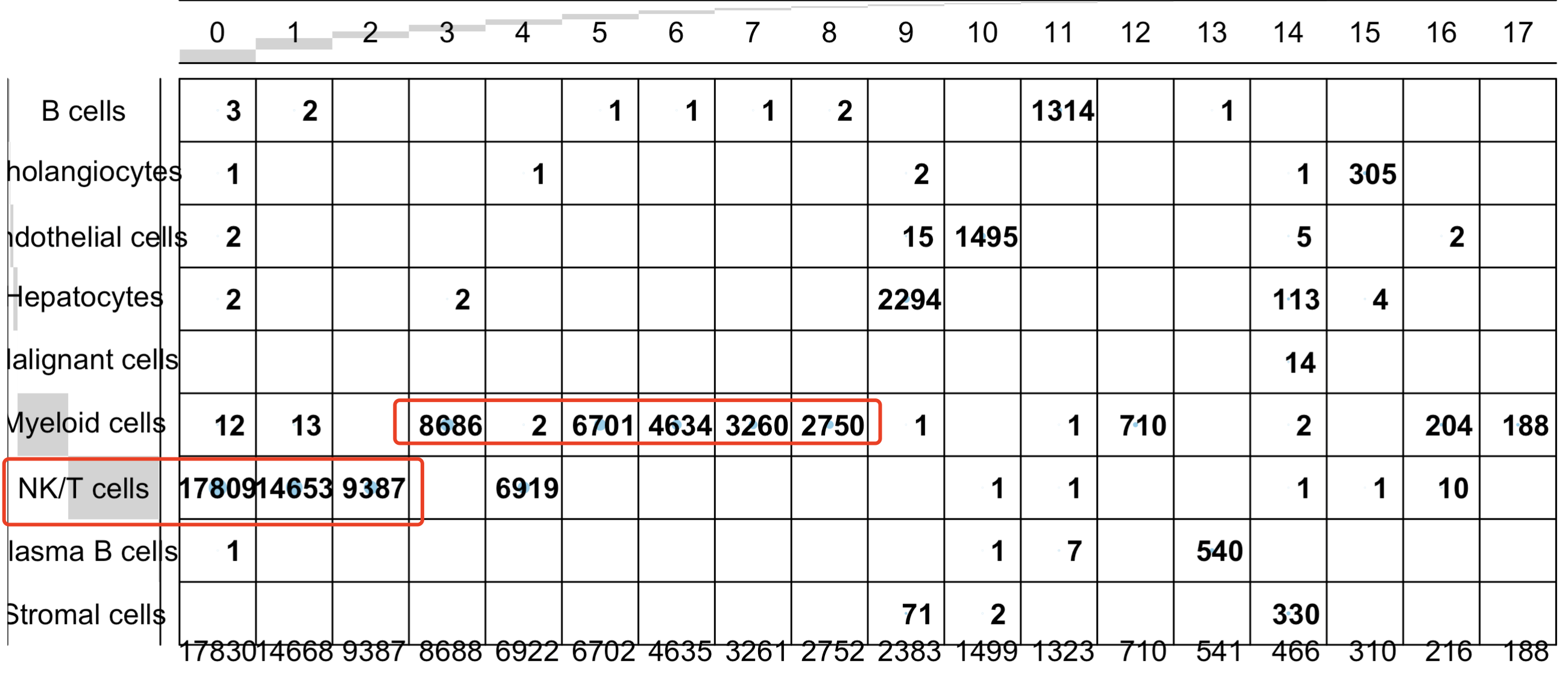
比如上面的顺序编号的0,1,2,4都是t或者nk细胞,但是它们在第一层次降维聚类分群的UMAP的二维坐标是很难有清晰界限的。也就是说 细分亚群的时候,其实是需要重新跑降维聚类分群了在每个子集细胞亚群里面:
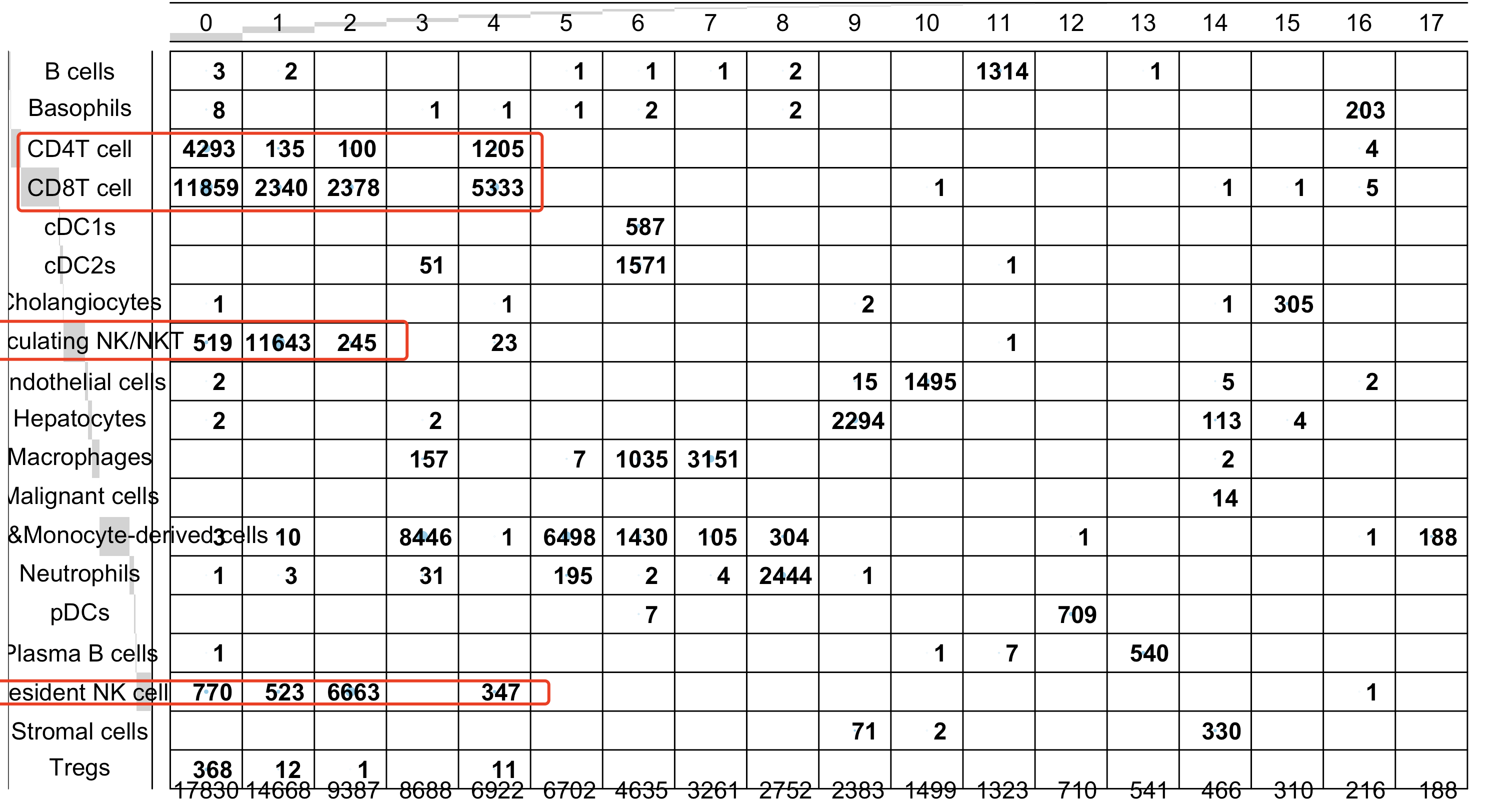
这个时候如果是我们想看看我们的第一层次降维聚类分群和第二层次降维聚类分群有什么对应关系,主要是淋巴细胞和髓系免疫细胞的细分:
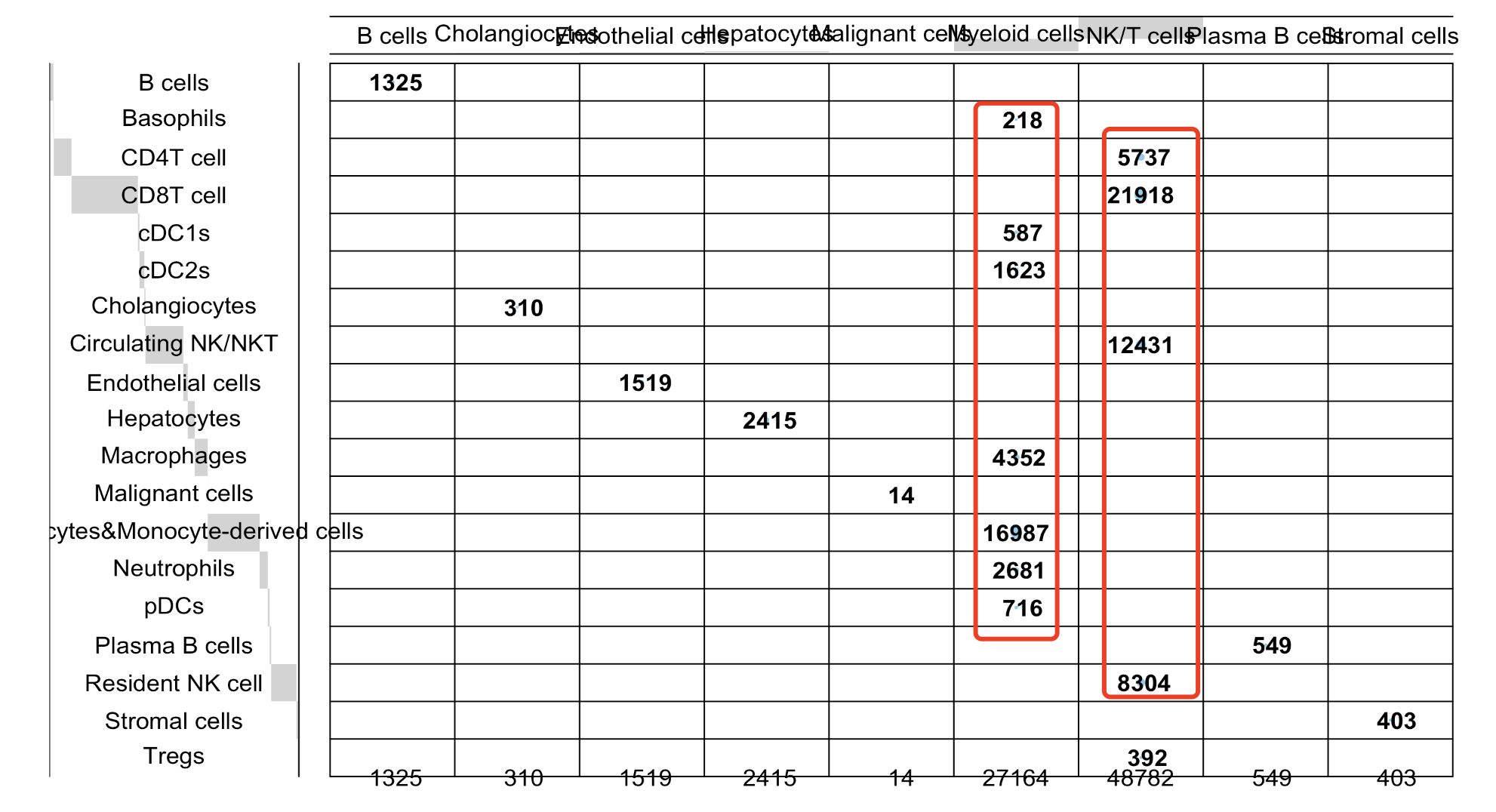
但是上面的可视化并不好看,我试试看想把它换成冲击图,代码如下所示:
plotdf=sce.all.int@meta.data[,c('Celltype.major','Celltype.minor')]
gplots::balloonplot(
table(plotdf$Celltype.major,
plotdf$Celltype.minor)
)
#桑基图
library(ggalluvial)
library(dplyr)
result <- plotdf %>%
count(Celltype.major, Celltype.minor)
head(result)
p<-ggplot(data = result,
aes(axis1 = Celltype.major, axis2 = Celltype.minor, y = n)) +
geom_alluvium(aes(fill = Celltype.major)) +
geom_stratum() +
geom_text(stat = "stratum", aes(label = after_stat(stratum))) +
theme_minimal()
p
发现也不算好看,一个简单的解决方案就是仅仅是保留淋巴细胞和髓系免疫细胞即可,因为只有它们才进行了细分亚群。
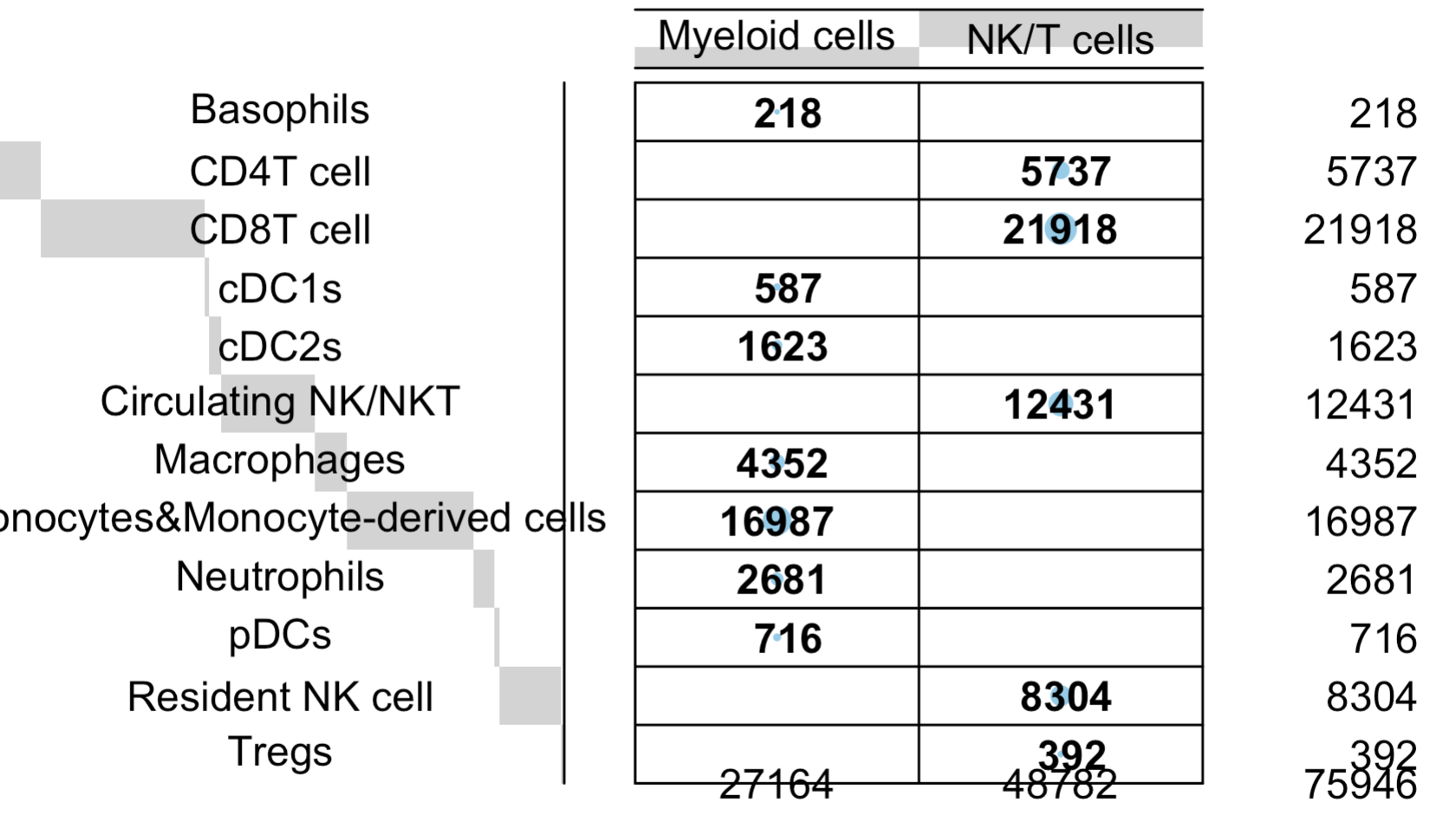
是否有其它更好的展示方式呢?