今天的单细胞天地公众号分享了:scanpy怎么分开读取GEO数据库的10X单细胞3个文件,使用Python的scanpy分别读取3个文件后,再组合起来。在R也是可以这样操作,但是会很麻烦。之前在在单细胞天地教程:表达矩阵逆转为10X的标准输出3个文件,详细介绍过 10X文件的3个标准文件。比如SRR7722939数据集里面,文件barcodes.tsv 和 genes.tsv,就是表达矩阵的行名和列名:
jmzengdeMacBook-Pro:SRR7722939 jmzeng$ head barcodes.tsv
AAACCTGAGCGAAGGG-1
AAACCTGAGGTCATCT-1
AAACCTGAGTCCTCCT-1
AAACCTGCACCAGCAC-1
AAACCTGGTAACGTTC-1
AAACCTGGTAAGGATT-1
AAACCTGGTTGTCGCG-1
AAACCTGTCCTGCCAT-1
AAACGGGAGTCATCCA-1
AAACGGGCATGGATGG-1
jmzengdeMacBook-Pro:SRR7722939 jmzeng$ head genes.tsv
hg38_ENSG00000243485 hg38_RP11-34P13.3
hg38_ENSG00000237613 hg38_FAM138A
hg38_ENSG00000186092 hg38_OR4F5
hg38_ENSG00000238009 hg38_RP11-34P13.7
hg38_ENSG00000239945 hg38_RP11-34P13.8
hg38_ENSG00000239906 hg38_RP11-34P13.14
hg38_ENSG00000241599 hg38_RP11-34P13.9
hg38_ENSG00000279928 hg38_FO538757.3
hg38_ENSG00000279457 hg38_FO538757.2
hg38_ENSG00000228463 hg38_AP006222.2
但是matrix.mtx,就稍微复杂一点,仔细看:
jmzengdeMacBook-Pro:SRR7722939 jmzeng$ head matrix.mtx
%%MatrixMarket matrix coordinate integer general
%
33694 2049 1878957
28 1 1
55 1 2
59 1 1
60 1 1
62 1 1
78 1 2
111 1 1
如果你关注这3个文件的行数:
2049 barcodes.tsv
33694 genes.tsv
1878960 matrix.mtx
就会发现,matrix.mtx文件里面的33694、2049、1878957数值,分别是细胞数量,基因数量,以及有表达量的值的数量(全部的值应该是33694X2049接近7000万,但是有值的仅仅是不到200万,所以单细胞矩阵里面只有3%左右的值大于0 )。每个10X样本都是走流程拿到10x单细胞转录组数据的3个文件的表达矩阵。
也就是说,我们拿到了的每个单细胞转录组项目的每个样品的文件名字通常是不符合规则,比如2022-GSE201048-羊癫疯患者大脑,如下所示:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE201048
每个样品的文件名字前面有前缀:
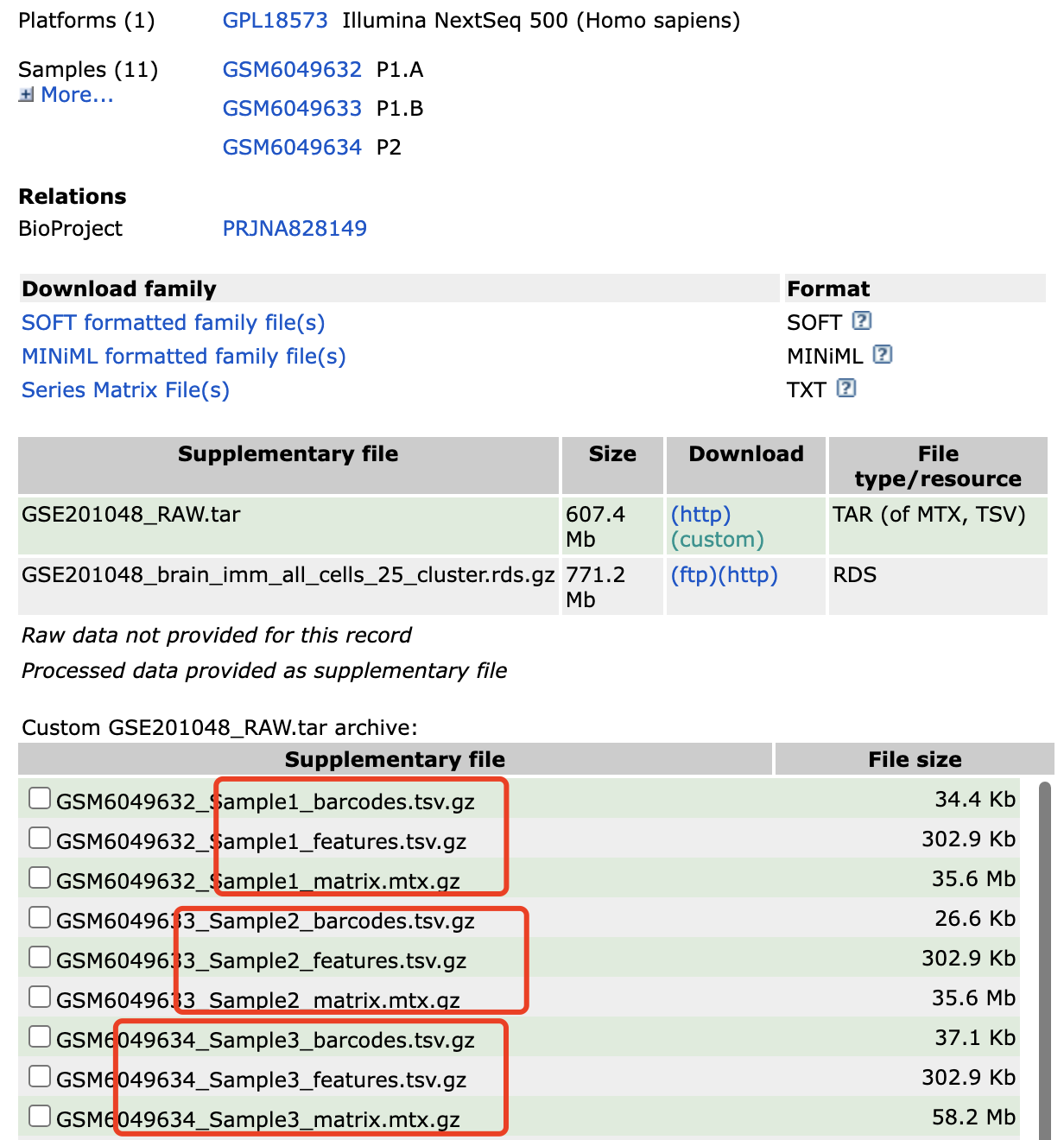
规则每个样品应该是有一个独立的文件夹,而文件夹里面的才是样品的3文件,而且文件名字需要是稳定的:文件名为 features.tsv.gz、matrix.mtx.gz 和 barcodes.tsv.gz。 不能有自定义的稀奇古怪的名字!所以我们分享了一个简单的r代码整理它,如下所示:
fs=list.files('./','features.tsv.gz')
fs
samples1=gsub('_features.tsv.gz','',fs)
samples1
library(stringr)
samples2=str_split(samples1,'_',simplify = T)[,1]
samples2 =samples1
length(unique(samples2))
samples2
#samples2 = samples1
lapply(1:length(samples2), function(i){
x=samples2[i]
y=samples1[i]
dir.create(x,recursive = T)
file.copy(from=paste0(y,'_features.tsv.gz'),
to=file.path(x, 'features.tsv.gz' ))
file.copy(from=paste0(y,'_matrix.mtx.gz'),
to= file.path(x, 'matrix.mtx.gz' ) )
file.copy(from=paste0(y,'_barcodes.tsv.gz'),
to= file.path(x, 'barcodes.tsv.gz' ))
})
然后让chatGPT帮我解释了一下:

最后的结果很完美,每个单细胞样品都是一个独立的文件夹,然后每个文件夹里面都是 文件名为 features.tsv.gz、matrix.mtx.gz 和 barcodes.tsv.gz的3个文件,接下来就很容易读取啦!
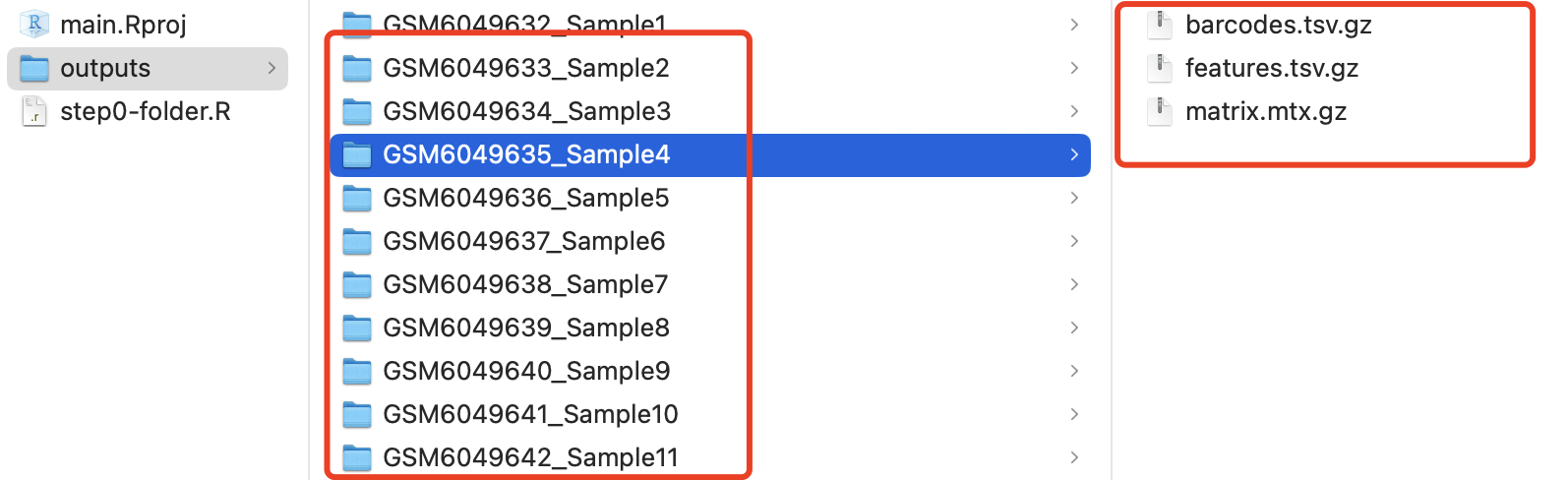
读取这个单细胞转录组项目的代码是:
rm(list=ls())
options(stringsAsFactors = F)
source('scRNA_scripts/lib.R')
getwd()
###### step1: 导入数据 ######
dir='GSE201048_RAW/outputs/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
tmp = Read10X(file.path(dir,pro ))
if(length(tmp)==2){
ct = tmp[[1]]
}else{ct = tmp}
sce =CreateSeuratObject(counts = ct ,
project = pro ,
min.cells = 5,
min.features = 300 )
return(sce)
})
do.call(rbind,lapply(sceList, dim))
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples )
names(sce.all@assays$RNA@layers)
sce.all[["RNA"]]$counts
# Alternate accessor function with the same result
LayerData(sce.all, assay = "RNA", layer = "counts")
sce.all <- JoinLayers(sce.all)
dim(sce.all[["RNA"]]$counts )
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all$orig.ident)
library(stringr)
sce.all$orig.ident=str_split(colnames(sce.all),'[-_]',simplify = T)[,2]
table(sce.all$orig.ident)
sce.all