早期(可能是五六年前)我们的单细胞转录组数据分析教程确实是提到过singleR的方法,它可以依赖于singleR自己的数据库文件去自动化注释单细胞转录组亚群。
但是因为singleR的数据库资源陈旧而且很有限,满足不了日益增长的单细胞应用,后面我们都是主推第一层次降维聚类分群后的人工命名,通常我们拿到了肿瘤相关的单细胞转录组的表达量矩阵后的第一层次降维聚类分群通常是:
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibro or CD31+,PECAM1,endo)
参考我前面介绍过 CNS图表复现08—肿瘤单细胞数据第一次分群通用规则,这3大单细胞亚群构成了肿瘤免疫微环境的复杂。绝大部分文章都是抓住免疫细胞亚群进行细分,包括淋巴系(T,B,NK细胞)和髓系(单核,树突,巨噬,粒细胞)的两大类作为第二次细分亚群。但是也有不少文章是抓住stromal 里面的 fibro 和endo进行细分,并且编造生物学故事的。
这样的话,因为强烈依赖于人工审查对单细胞亚群进行生物学命名,所以工作量很大。前面我们已经介绍了心肝脾肺肾等多个器官的上皮细胞的细分亚群, 以及免疫细胞里面的髓系和B细胞细分亚群:
目前就是stromal 里面的 fibro 和endo进行细分我还介绍的不够,其实stromal 里面不仅仅是 fibro 和endo,还有周细胞和SMC,不过大多数情况下肿瘤样品里面的基质细胞并不多,所以不一定能细分清楚。但是如果专门的取基质细胞的,就很清晰可以看到了,比如2021的文章:《Resolving the intertwining of inflammation and fibrosis in human heart failure at single‐cell level》,数据集是GSE145154 ,里面 介绍很清晰的区分了 fibro 和endo以及周细胞和SMC:
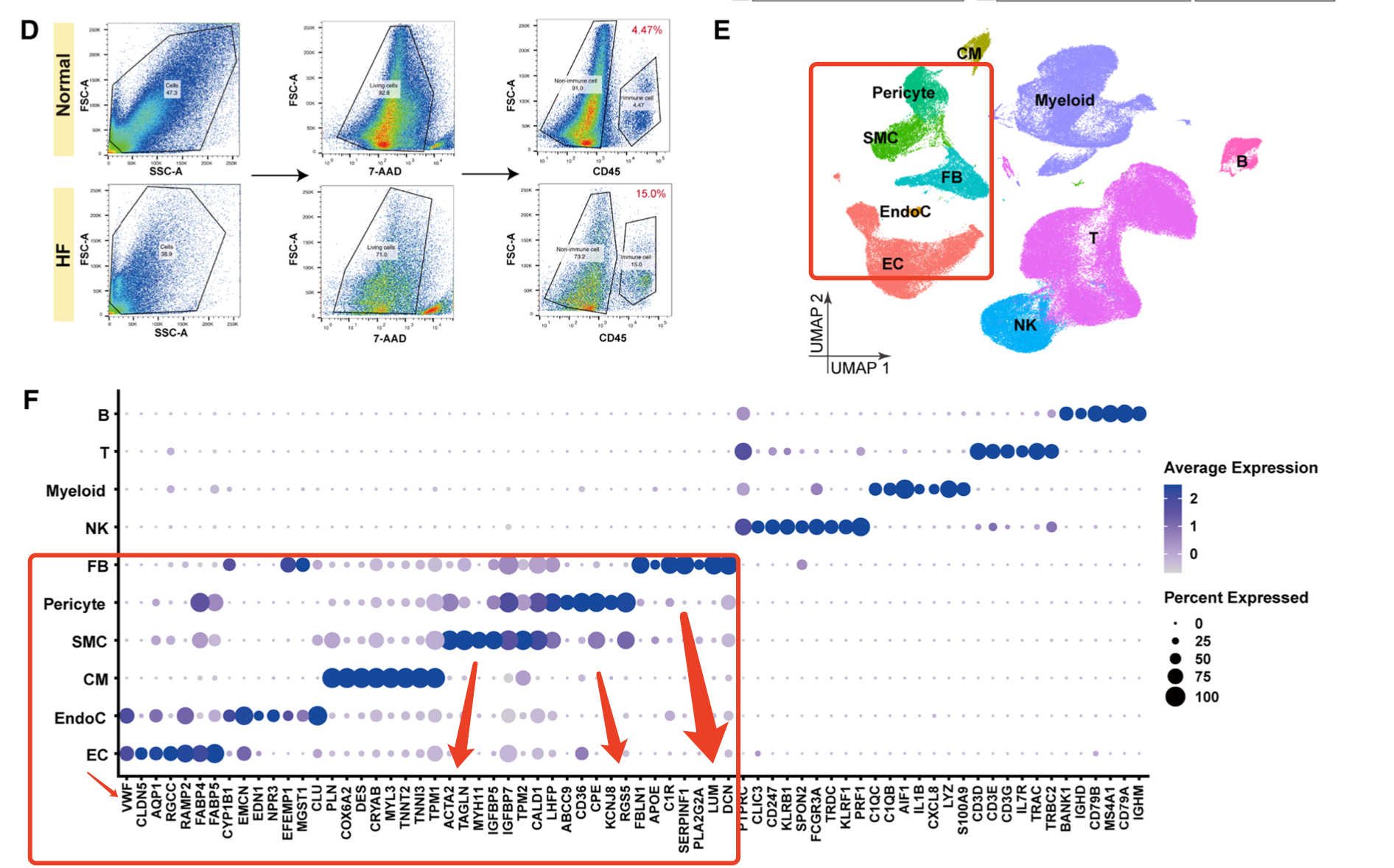
而且很明显,上面的细胞和SMC其实是会相互混入的,这个暂时是无解的。而且这样的多层次分群,很明显是超出了singleR的能力,因为singleR的数据库就没有这样的stromal的细分。但是singleR确实是有自己的优点,比如它可以不需要提前降维聚类分群,直接针对表达量矩阵本身,就可以给每个细胞一个身份,这样的话它就跳过了降维聚类分群过程引入的误差。如果仅仅是因为singleR的数据库资源的匮乏,就放弃这个工具未免有点暴殄天物。让我们来演示一下如何为singleR方法自己构建一个特殊生物学领域的数据库吧:
singleR的书籍
你能想象吗,singleR的知识点细节都足以写成一本书了,详见:https://bioconductor.org/books/release/SingleRBook/
Authors: Aaron Lun [aut, cre]
Version: 1.12.1
Modified: 2023-11-29
Compiled: 2023-12-06
Environment: R version 4.3.2 Patched (2023-11-13 r85521), Bioconductor 3.18
License: CC BY
Copyright: Aaron Lun, 2020
Source: https://github.com/LTLA/SingleRBook-base
但是我们只需要看这个书籍的第一个章节的1.3单元内容即可,是一个Quick start的案例演练,拿了pbmc4k这样的单细胞转录组数据集作为案例,然后使用singleR自己的HumanPrimaryCellAtlasData数据库文件来进行注释,如下所示:
# Loading test data.
library(TENxPBMCData)
new.data <- TENxPBMCData("pbmc4k")
# Loading reference data with Ensembl annotations.
library(celldex)
ref.data <- HumanPrimaryCellAtlasData(ensembl=TRUE)
# Performing predictions.
library(SingleR)
predictions <- SingleR(test=new.data, assay.type.test=1,
ref=ref.data, labels=ref.data$label.main)
table(predictions$labels)
可以看到它需要的是一个数据库文件,然后只需要使用SingleR包里面的SingleR函数即可把数据库里面的细胞亚群注释信息映射到需要命名的单细胞转录组数据集里面。
成功的运行了SingleR包里面的SingleR函数之后,就可以拿到每个单细胞的具体的身份信息,如下所示:
##
## B_cell CMP DC GMP
## 606 8 1 2
## Monocyte NK_cell Platelets Pre-B_cell_CD34-
## 1164 217 3 46
## T_cells
## 2293
现在的问题是我们不满意singleR自己的HumanPrimaryCellAtlasData数据库文件,所以需要自己构建一个类似的信息文件。
singleR有一个官方数据库资源包celldex
singleR有一本书的知识点也就算了,它还把自己的官方数据库资源做成了一个包celldex,值得注意是 Each dataset contains a log-normalized expression matrix that is intended to be comparable to log-UMI counts from common single-cell protocols (Aran et al. 2019) or gene length-adjusted values from bulk datasets.
也就是说,它的每个数据库本质上就是一个bulk转录组测序后的表达量矩阵,获取的方法,如下所示:
# BiocManager::install("celldex")
library(celldex)
ref <- HumanPrimaryCellAtlasData()
ref <- BlueprintEncodeData()
ref <- MouseRNAseqData()
ref <- ImmGenData()
ref <- DatabaseImmuneCellExpressionData()
ref <- NovershternHematopoieticData()
ref <- MonacoImmuneData()
说实话,这个官方数据库资源包celldex里面的7大数据库资源,都不好用。这也就是为什么我们最近五六年的单细胞转录组经常补在推荐这个singleR方法学了,但是确实是可以通过自建一个数据库来避开它的缺点。
数据库信息来源
自建数据库,通常指的是我们已经做好了的单细胞转录组降维聚类分群结果,而且成功拿到了亚群名字,比如这个2020的文章:《A Single-Cell Atlas of the Human Healthy Airways》,它就拿到了如下所示的结果:
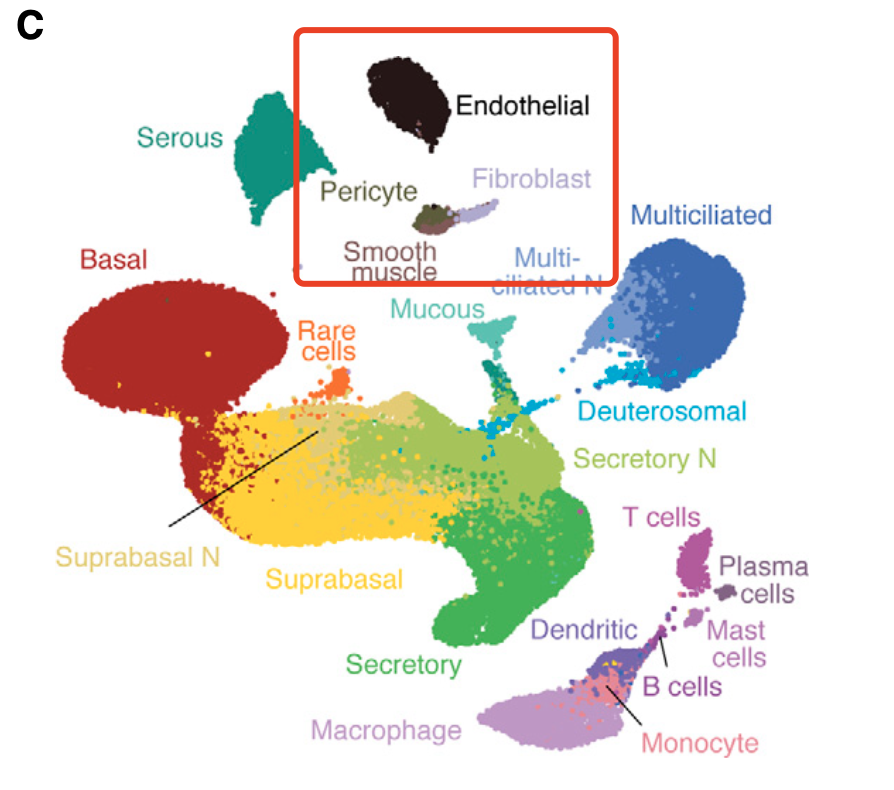
可以看到,里面确实是有 fibro 和endo以及周细胞和SMC 信息,我们读取文章的公开信息:web tool ( https://www.genomique.eu/cellbrowser/HCA/ ). 这里面有表达量矩阵文件以及配套的细胞亚群名字信息。
ct=fread( 'Raw_exprMatrix.tsv.gz',data.table = F)
ct[1:4,1:4]
rownames(ct)=ct[,1]
ct=ct[,-1]
sce.all =CreateSeuratObject(counts = ct ,
min.cells = 5,
min.features = 300 )
理解常规的单细胞转录组降维聚类分群代码,可以看 链接: https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh ,基本上大家只需要读入表达量矩阵文件到r里面就可以使用Seurat包做全部的流程。上面我们下载了 Raw_exprMatrix.tsv.gz 文件然后读取后构建了Seurat对象,接下来就读取细胞亚群注释信息文件 ,然后选取我们需要的单细胞亚群:
phe2=data.table::fread('supplements/meta.tsv',data.table = F)
rownames(phe2)=phe2$Id
sce.all.int = sce.all
phe=sce.all.int@meta.data
head(phe)
ids=intersect(rownames(phe),rownames(phe2))
chooseCT = c('Fibroblast','Smooth muscle','Pericyte','Endothelial',
'Macrophage','Monocyte','Dendritic','LT/NK','B cells','Plasma cells')
tmp = phe2[ids,]
tmp = tmp[tmp$CellType %in% chooseCT,]
sce.singleR=sce.all.int[,colnames(sce.all.int) %in% tmp$Id]
sce.singleR$paper = tmp[match(colnames(sce.singleR),tmp$Id),'CellType']
DimPlot(sce.singleR,group.by = 'paper',label = T,repel = T)
save(sce.singleR,file = 'sce.singleR.Rdata')
as.data.frame(sort(table(sce.singleR$paper)))
得到如下所示的结果:
Var1 Freq
1 B cells 11
2 Plasma cells 19
3 Smooth muscle 35
4 Pericyte 45
5 Fibroblast 49
6 Dendritic 63
7 Monocyte 76
8 LT/NK 98
9 Macrophage 305
10 Endothelial 398
可以看到的是这个文件会很小,因为细胞数量确实是不多,但是已经是有 fibro 和endo以及周细胞和SMC 信息,以及部分免疫细胞亚群信息。
构建适配singleR算法的数据库文件
前面拿到了一个Seurat单细胞转录组对象,里面有10个单细胞亚群,但是它并不是SingleR包里面的SingleR函数需要的格式(SingleCellExperiment)。可以做一个简单的转:
load(file = 'sce.singleR.Rdata')
sce = sce.singleR
colnames(sce@meta.data)
Idents(sce) = sce$paper
table(Idents(sce) )
##NOTE:以前是AverageExpression
av <-AggregateExpression(sce , # group.by = "celltype",
assays = "RNA")
Ref = av[[1]]
head(Ref)
ref_sce=SingleCellExperiment::SingleCellExperiment(assays=list(counts=Ref))
library(scater)
ref_sce=scater::logNormCounts(ref_sce)
library(SingleCellExperiment)
logcounts(ref_sce)[1:4,1:4]
colData(ref_sce)$Type=colnames(Ref)
table(colnames(Ref))
ref_sce
save(ref_sce,file = 'HCA_airway_ref_sce.Rdata')
有了上面的 ref_sce 变量,就可以使用SingleR包里面的SingleR函数啦。
然后处理需要做注释的单细胞转录组数据集
我们这里举例的文章是2020发表在NC的:《Single-cell transcriptome atlas of the human corpus cavernosum》,对应的数据集是GSE206528,可以看到里面的9个单细胞转录组样品的表达量矩阵文件如下所示:
21M 6 21 2022 GSM6255907_Normal_1_gene_martix.csv.gz
16M 6 21 2022 GSM6255908_Normal_2_gene_matrix.csv.gz
27M 6 21 2022 GSM6255909_Normal_3_gene_matrix.csv.gz
16M 6 21 2022 GSM6255910_non-DM1_gene_matrix.csv.gz
9.6M 6 21 2022 GSM6255911_non-DM2_gene_matrix.csv.gz
28M 6 21 2022 GSM6255912_non-DM3_gene_matrix.csv.gz
25M 6 21 2022 GSM6255913_Diabetes_1_gene_matrix.csv.gz
26M 6 21 2022 GSM6255914_Diabetes_2_gene_martix.csv.gz
这个GSE206528的单细胞转录组数据集,很容易构建成为Seurat对象。仍然是走常规的单细胞转录组降维聚类分群代码,可以看 链接: https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh ,基本上大家只需要读入表达量矩阵文件到r里面就可以使用Seurat包做全部的流程,这个时候需要自己肉眼去看每个亚群标记基因,然后才有可能去给具体的单细胞亚群一个生物学名字:
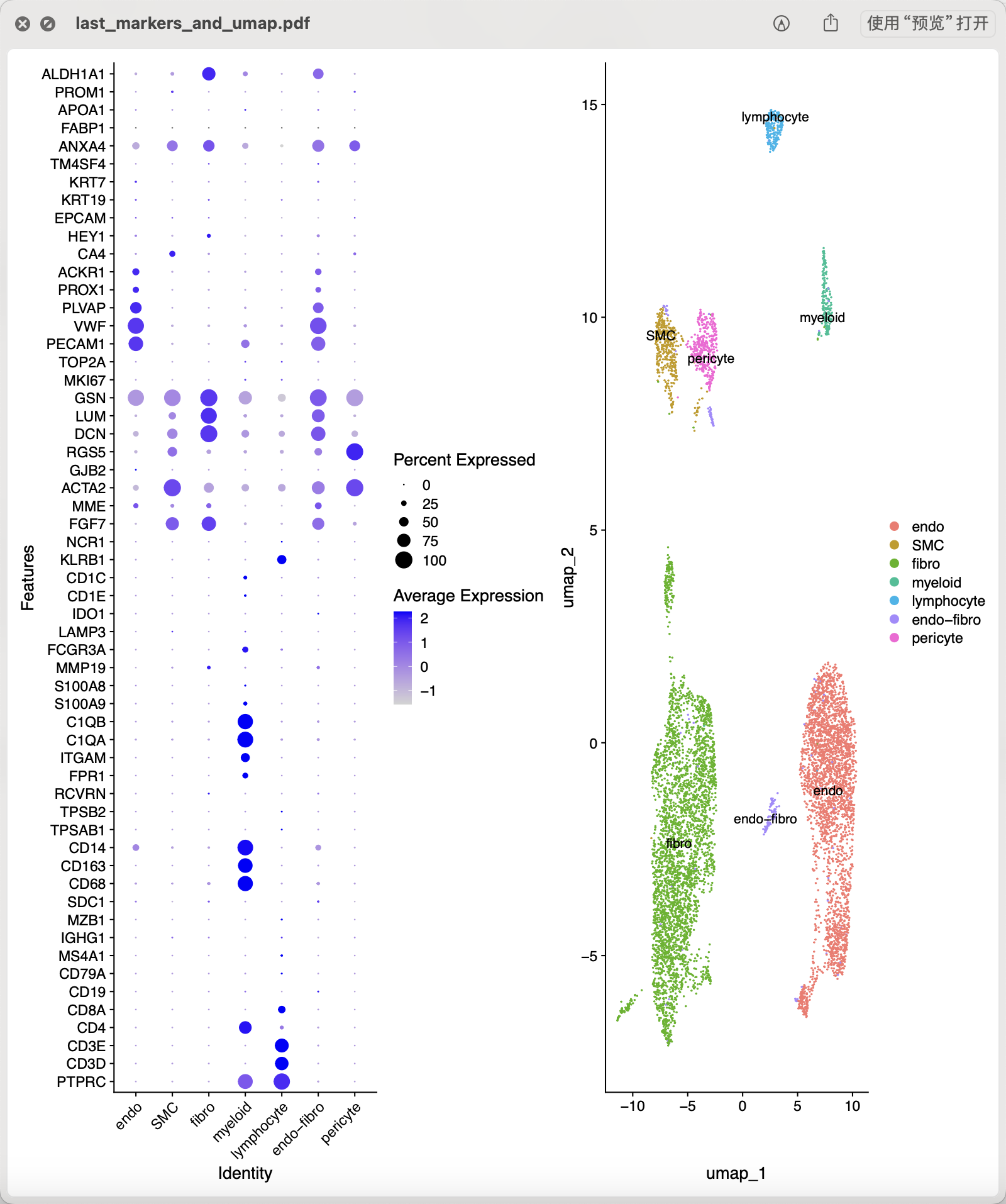
而且可以看到,上面的淋巴系(T,B,NK细胞)和髓系(单核,树突,巨噬,粒细胞)的两大类在我们的第一层次降维聚类分群里面其实是只能说是分成两个大类,没办法细分里面的具体亚群。
但是,如果使用SingleR包里面的SingleR函数,其实是可以跨越上面的常规的降维聚类分群,直接使用单细胞表达量矩阵本身即可:
load('./singleR/HCA_airway_ref_sce.Rdata')
ref_sce
source('scRNA_scripts/lib.R')
ce.all = readRDS('2-harmony/sce.all_int.rds')
testdata <- GetAssayData(sce.all, slot="data")
接下来,运行 SingleR包里面的SingleR函数 是很简单的事情;
library(SingleR)
pred <- SingleR(test=testdata, ref=ref_sce,
labels=ref_sce$Type
)
as.data.frame(table(pred$labels))
head(pred)
labels=pred$labels
table(labels)
save(labels,file = 'SingleR_celltype.Rdata')
sce.all$singleR=labels
DimPlot(sce.all, reduction = "umap",repel = T,
group.by = "singleR",label = T)
ggsave('umap-by-singleR.pdf',width = 8,height = 6)
可以看到,效果还不错:

而且第一层次降维聚类分群里面的淋巴系(T,B,NK细胞)和髓系(单核,树突,巨噬,粒细胞)的两大类在SingleR包里面的SingleR函数效果下面也细分的很清楚。
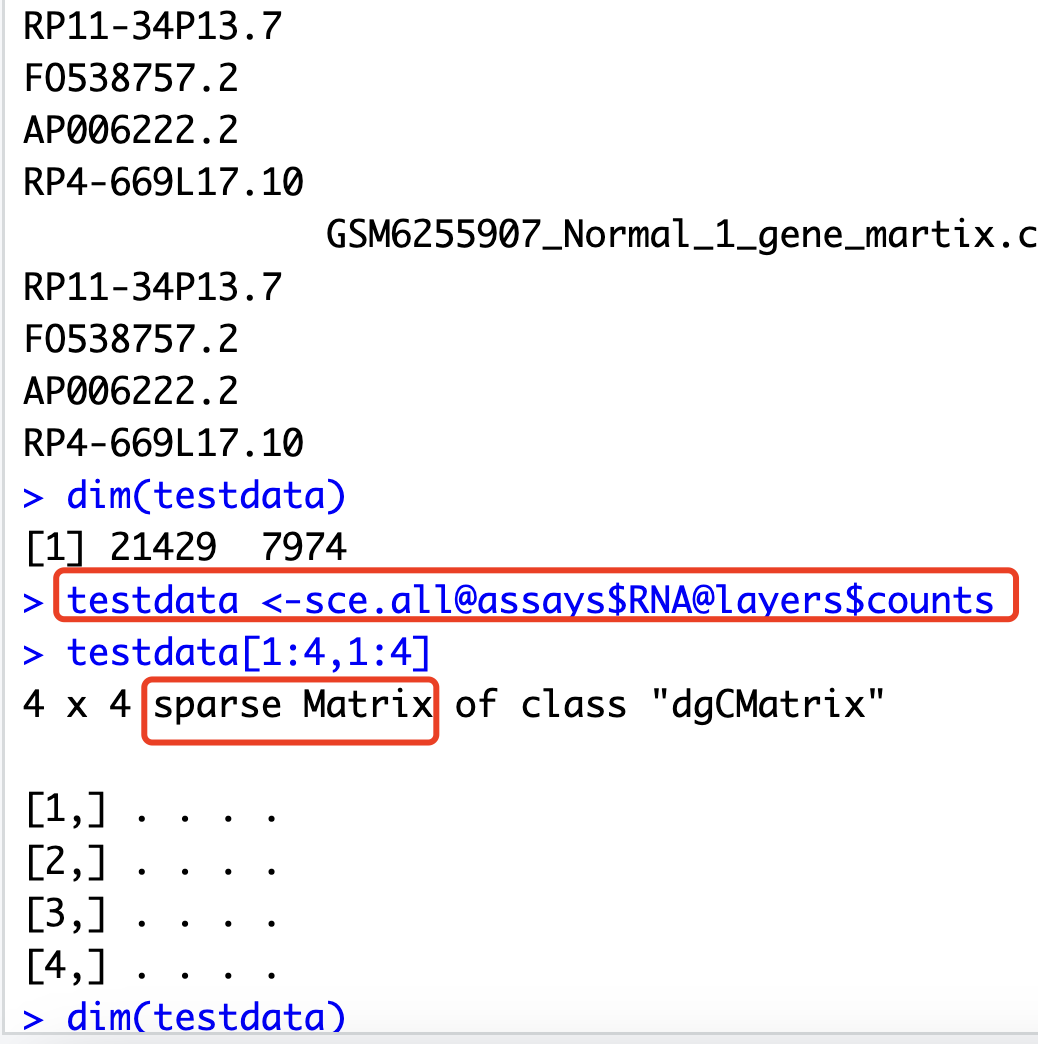
另外,值得注意的是,如果我们想从Seurat对象里面获取表达量矩阵,有3个不一样的代码;
testdata <- GetAssayData(sce.all, slot="data")
testdata <-sce.all@assays$RNA$counts
testdata <-sce.all@assays$RNA@layers$counts
testdata[1:4,1:4]
dim(testdata)
就是最后一个方法获取的是没有基因名字和细胞名字的稀疏矩阵,蛮有意思的:
学徒作业
前面提到的2021的文章:《Resolving the intertwining of inflammation and fibrosis in human heart failure at single‐cell level》,数据集是GSE145154 ,里面 介绍很清晰的区分了 fibro 和endo以及周细胞和SMC。大家试试看常规降维聚类分群后的命名,和我们演示的SingleR包里面的SingleR函数的效果,对比一下。