单细胞转录组数据分析过程有几个难点:
- 去除双细胞
- 去除低质量细胞亚群
- 回归掉细胞周期等因素
实际上初学者完全不需要使用各种花里胡哨的工具来完成上面的步骤,因为所有的细节都在降维聚类分群结果里面看得到!让我们来演练一下:
比如数据集GSE217845,从里面挑选出来10个单细胞转录组样品,都是10x技术的,如下所示 :
一般来说,作者上传的10x的3文件都是过滤好的了,这个时候理论上10x技术会给每个样品产出10000个左右的细胞,我们做一个简单的批量读取即可;
# 参考:https://mp.weixin.qq.com/s/tw7lygmGDAbpzMTx57VvFw
dir='outputs/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
tmp = Read10X(file.path(dir,pro ))
if(length(tmp)==2){
ct = tmp[[1]]
}else{ct = tmp}
print(dim(ct))
sce =CreateSeuratObject(counts = ct ,
project = pro ,
min.cells = 5,
min.features = 300 )
return(sce)
})
do.call(rbind,lapply(sceList, dim))
可以看到的是这个时候其实已经引入了第一个过滤流程,就是 min.cells = 5, min.features = 300 这两个参数,这个时候是一个非常初步的质量控制,是可以更严格的,比如提高 min.features = 500 甚至更高。不过先我们看看初步质量控制的效果吧:
> do.call(rbind,lapply(sceList, dim))
[,1] [,2]
[1,] 18742 14073
[2,] 20394 2758
[3,] 20273 6565
[4,] 18243 7600
[5,] 19946 14152
[6,] 18571 8436
[7,] 20354 9151
[8,] 21206 8856
[9,] 20526 15214
[10,] 19873 4076
如果大家自行看 min.cells = 5, min.features = 300 这两个参数 的效果,就会发现它仅仅是对每个样品都过滤了一千多个细胞。最后得到了 9万多个细胞的单细胞表达量矩阵;
> sce.all
An object of class Seurat
23757 features across 90881 samples within 1 assay
Active assay: RNA (23757 features, 0 variable features)
1 layer present: counts
常规的单细胞转录组降维聚类分群代码可以看 链接: https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh ,基本上大家只需要读入表达量矩阵文件到r里面就可以使用Seurat包做全部的流程!
先跑默认流程
我们默认的过滤流程里面是:
#根据上述指标,过滤低质量细胞/基因
#过滤指标1:最少表达基因数的细胞&最少表达细胞数的基因
# 一般来说,在CreateSeuratObject的时候已经是进行了这个过滤操作
# 如果后期看到了自己的单细胞降维聚类分群结果很诡异,就可以回过头来看质量控制环节
# 先走默认流程即可
selected_c <- WhichCells(input_sce, expression = nFeature_RNA > 500)
selected_f <- rownames(input_sce)[Matrix::rowSums(input_sce@assays$RNA$counts > 0 ) > 3]
input_sce.filt <- subset(input_sce, features = selected_f, cells = selected_c)
#过滤指标2:线粒体/核糖体基因比例(根据上面的violin图)
selected_mito <- WhichCells(input_sce.filt, expression = percent_mito < 25)
selected_ribo <- WhichCells(input_sce.filt, expression = percent_ribo > 3)
selected_hb <- WhichCells(input_sce.filt, expression = percent_hb < 1 )
走了上面的代码之后拿到的单细胞表达量矩阵是:
> sce.all.filt
An object of class Seurat
23757 features across 76347 samples within 1 assay
Active assay: RNA (23757 features, 0 variable features)
1 layer present: counts
可以看到是过滤了一万多的细胞,然后就可以降维聚类分群啦,如下所示的惨不忍睹的效果 :
并不是说没办法给出来细胞亚群的名字,标准代码会出很多图,比如里面的质量控制图就可以看出来10这个亚群就属于低质量的,说明我们的过滤标准是不够严格的。

然后自行探索最佳质量控制流程
我们做了一个简单的探索:
sce.all.filt=subset(sce.all.filt,subset = percent_mito < 40 &
nCount_RNA <10000 &
nFeature_RNA <7500&
nFeature_RNA>500)
sce.all.filt
> sce.all.filt
An object of class Seurat
23757 features across 50292 samples within 1 assay
Active assay: RNA (23757 features, 0 variable features)
1 layer present: counts
可以看到,如果是这个标准,最开始9万细胞到现在就只剩下了5万啦。上面的最低阈值就去除了低质量细胞,最高阈值就去除了双细胞,当然了肯定是会错杀一批。但是数据分析其实很难得到百分百准确的结果,关键是我们如何合理的展示和解释我们的结果。
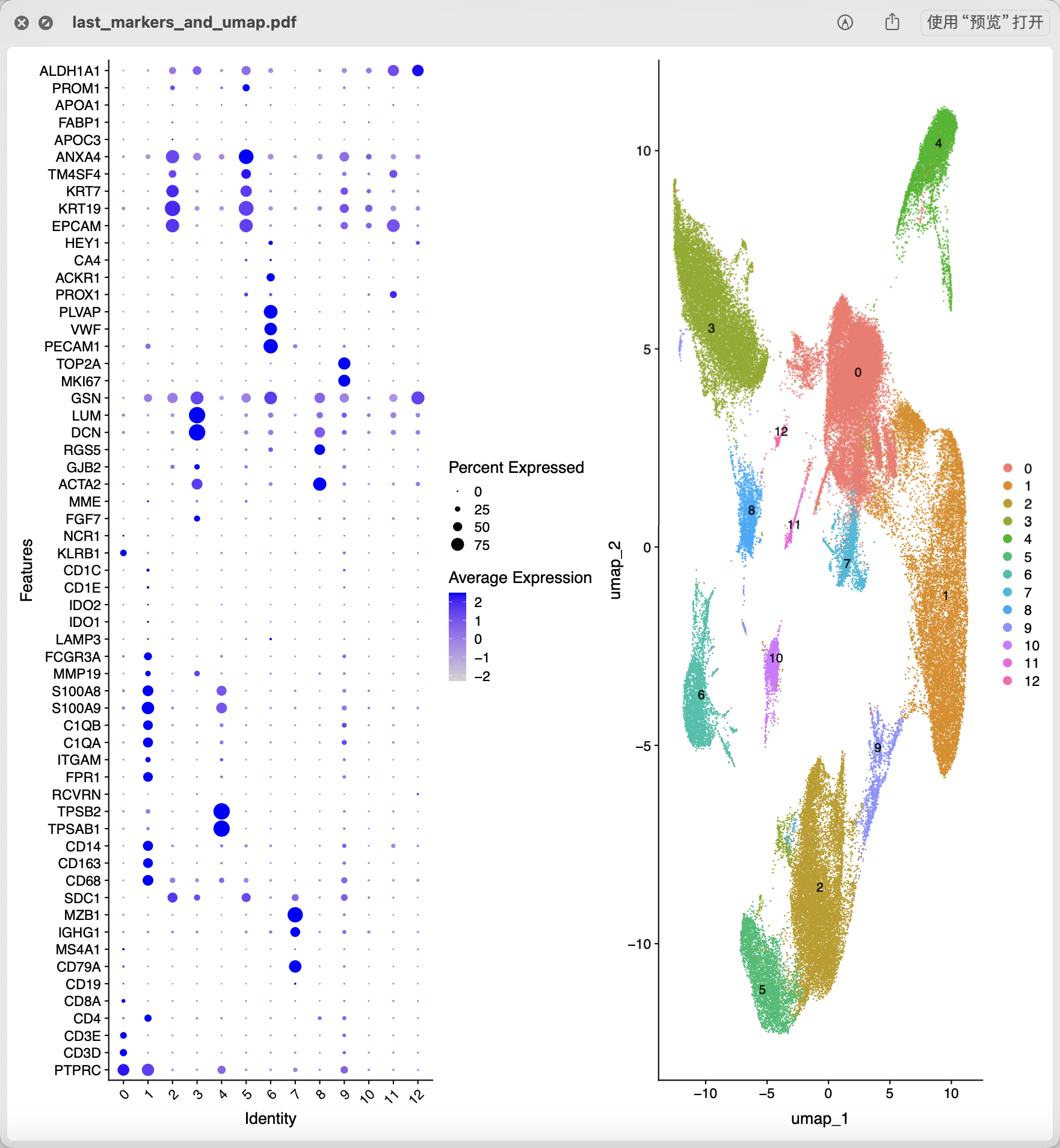
实是很严格,而且后面的降维聚类分群效果好很多啦!各个生物学功能亚群之间的距离是很清晰的,而且是能够给出来名字的!而且可以很清楚的看到细胞增殖相关单细胞亚群也不见了,虽然我们并没有去回归掉这个因素的影响。而且哪怕是我们结果里面仍然是有细胞增殖相关单细胞亚群也完全无需担心啊,你如实的命名它为细胞增殖相关单细胞亚群即可!

可以看到,上图里面的标号为2的亚群就是髓系免疫细胞亚群,通常我们拿到了肿瘤相关的单细胞转录组的表达量矩阵后的第一层次降维聚类分群通常是:
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibro or CD31+,PECAM1,endo)
参考我前面介绍过 CNS图表复现08—肿瘤单细胞数据第一次分群通用规则,这3大单细胞亚群构成了肿瘤免疫微环境的复杂。绝大部分文章都是抓住免疫细胞亚群进行细分,包括淋巴系(T,B,NK细胞)和髓系(单核,树突,巨噬,粒细胞)的两大类作为第二次细分亚群。但是也有不少文章是抓住stromal 里面的 fibro 和endo进行细分,并且编造生物学故事的。
学徒作业
理解常规的单细胞转录组降维聚类分群代码,可以看 链接: https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh ,基本上大家只需要读入表达量矩阵文件到r里面就可以使用Seurat包做全部的流程。
从上面的数据集GSE217845里面的10个胰腺癌的10x技术单细胞转录组数据的第一层次降维聚类分群里面提前髓系免疫细胞后,继续细分降维聚类拿到里面的巨噬细胞,然后继续细分巨噬细胞看看能否复现文章里面的:
