最近实习生在跑我布置的单细胞转录组练习题的时候汇报给我了一个很有意思的“视觉效应”,使用的仍然是最经典的数据集,是来自于SeuratData包的ifnb数据集,详见;SCTransform真的能完美替代Seurat早期的3个函数吗。代码如下所示;
rm(list = ls())
library(Seurat)
library(SeuratData)
library(ifnb.SeuratData)
data("ifnb")
ifnb=UpdateSeuratObject(ifnb)
table(ifnb$orig.ident)
table(ifnb$seurat_annotations,ifnb$orig.ident)
可以看到这个数据集是两个样品(两个处理)的单细胞转录组,而且是被提前注释完毕了的:
IMMUNE_CTRL IMMUNE_STIM
CD14 Mono 2215 2147
CD4 Naive T 978 1526
CD4 Memory T 859 903
CD16 Mono 507 537
B 407 571
CD8 T 352 462
T activated 300 333
NK 298 321
DC 258 214
B Activated 185 203
Mk 115 121
pDC 51 81
Eryth 23 32
这样的话,就很容易降维聚类分群,而且可以看到CD14和CD16的单核细胞很清晰 :
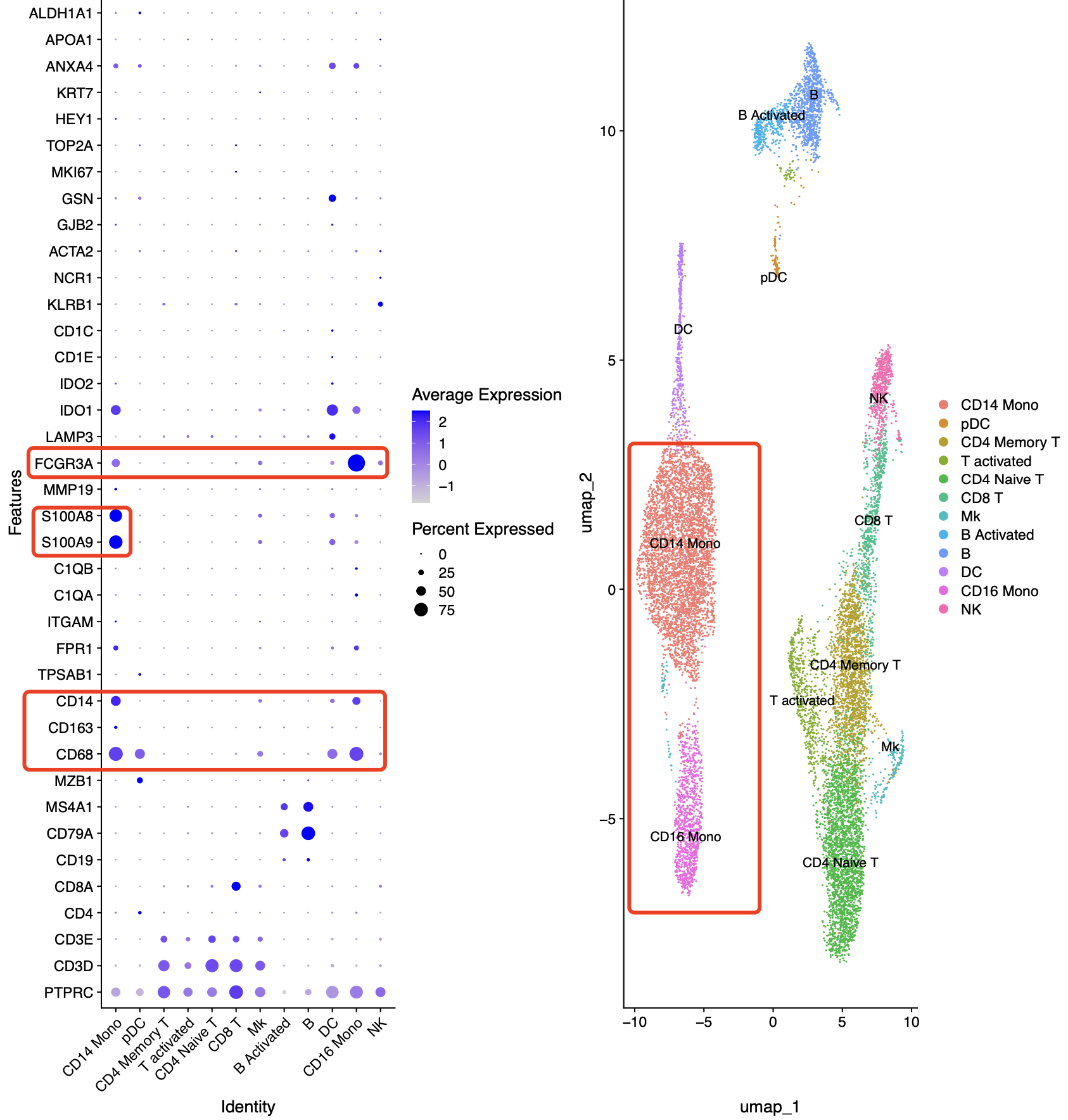
值得注意的是上面的代码是我们长久以来分享的标准流程哦,代码在:(链接: https://pan.baidu.com/s/1pKEnPmWXi-pTab0WZUWzgg?pwd=a7s1),里面的很多方法学和参数都是可以修改的, 但是如果修改就会面临不可控因素,详见;SCTransform真的能完美替代Seurat早期的3个函数吗。可以看到这个标准流程因为是全部的单细胞亚群的第一层次降维聚类分群,所以cd14其实是很难看出来有细分亚群或者说有样品差异,因为在第一层次降维聚类分群我们默认使用了harmony抹除了这个差异,但是如果你仔细的提取里面的单核细胞去细分亚群就可以看到内部的异质性哈。
因为我给学徒布置的任务就是看单核细胞的拟时序,所以他就做了一个很简单的可视化,就是使用DotPlot函数去可视化CD14和CD16的单核细胞的一些基因,如下所示的代码:
cg = c( 'CD68', 'CD163', 'CD14', 'FCGR3A',
'S100A9', 'S100A8', 'MMP19' )
p = DotPlot(ifnb ,cg,group.by = 'seurat_annotations' ,split.by = 'orig.ident')
p$data
吊轨的事情就出来了,理论上CD14和CD16的单核细胞的特异性基因不应该是受制于样品的,它应该是在两个样品(两个处理)的单细胞转录组,都应该是高表达量的,但是很明显下面就出现了在STIM组里面的远高于CTRL组的情况:
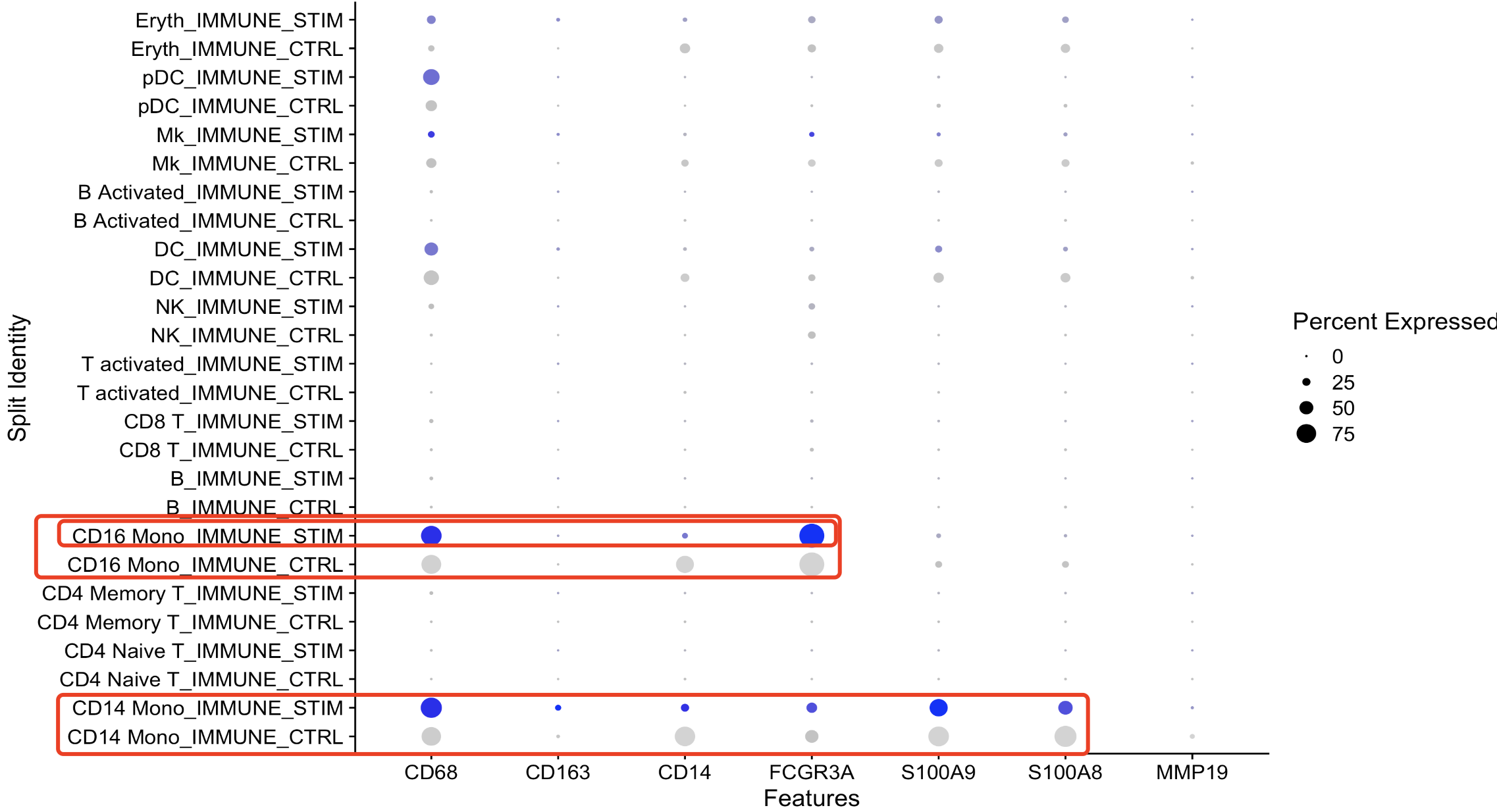
因为CD14和CD16的单核细胞的本质属性,它不应该是会低表达量,但是如果仅仅是看上图我们就会误认为单核细胞的标记基因在CTRL组的表达量都很低很低!
但是如果我们首先把这个单细胞转录组数据集按照两个样品(两个处理)拆分后再进行DotPlot函数去可视化CD14和CD16的单核细胞的一些基因,如下所示的代码:
DotPlot(ifnb[,ifnb$orig.ident=='IMMUNE_CTRL'],cg,group.by = 'seurat_annotations' )/
DotPlot(ifnb[,ifnb$orig.ident=='IMMUNE_STIM'],cg,group.by = 'seurat_annotations' )
可以看到在每个样品内部其实是:
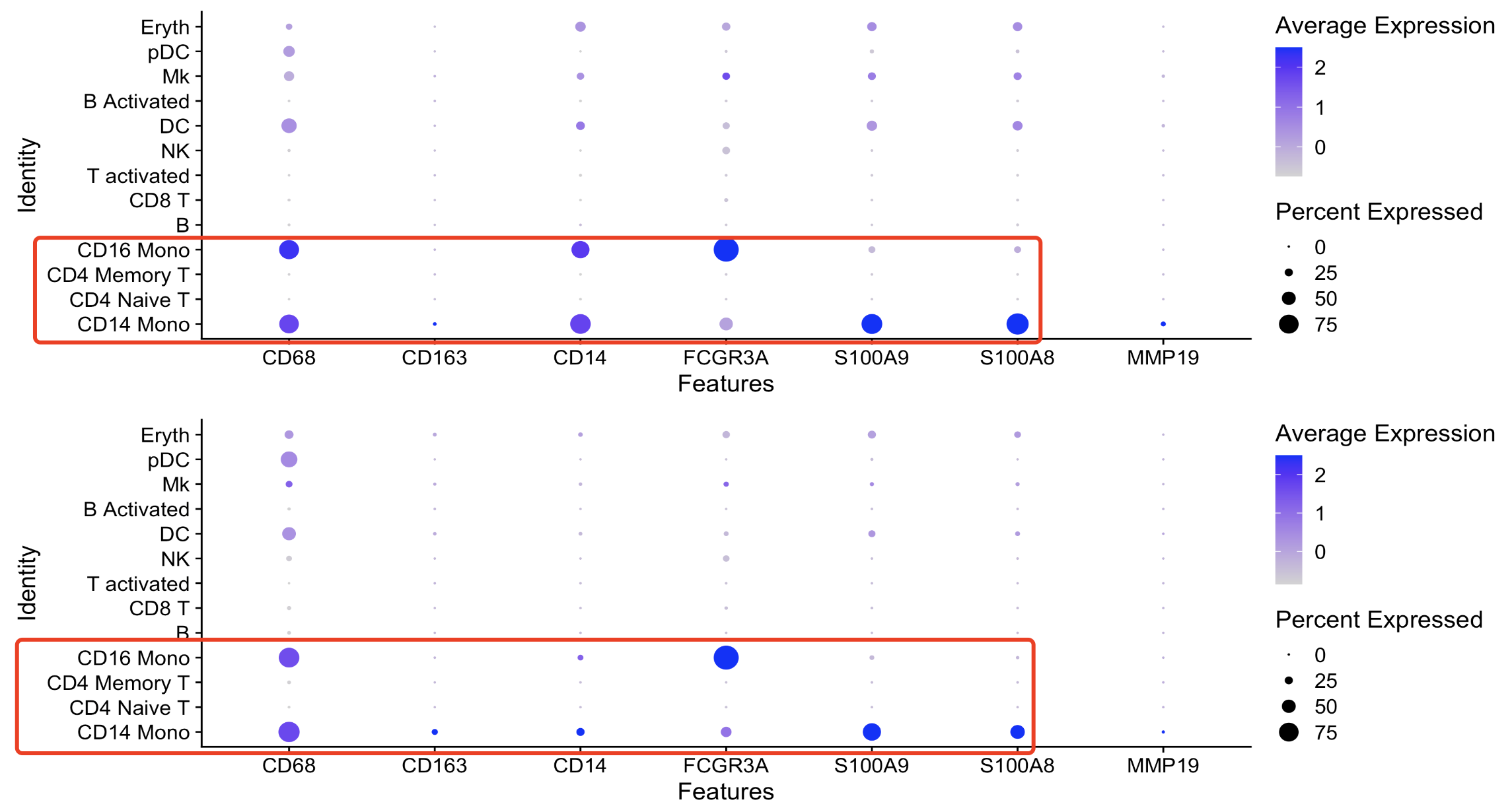
其实很容易理解它,就是可视化其实凸显的是相对概念,第一次可视化的时候如果我们肉眼看看 p$data 就可以发现,其实是CD16这个记忆在两个样品(两个处理)都是高表达,但是它因为在STIM组里面的远高于CTRL组,所以就相对表达量很低了,在DotPlot函数去可视化就好奇怪。
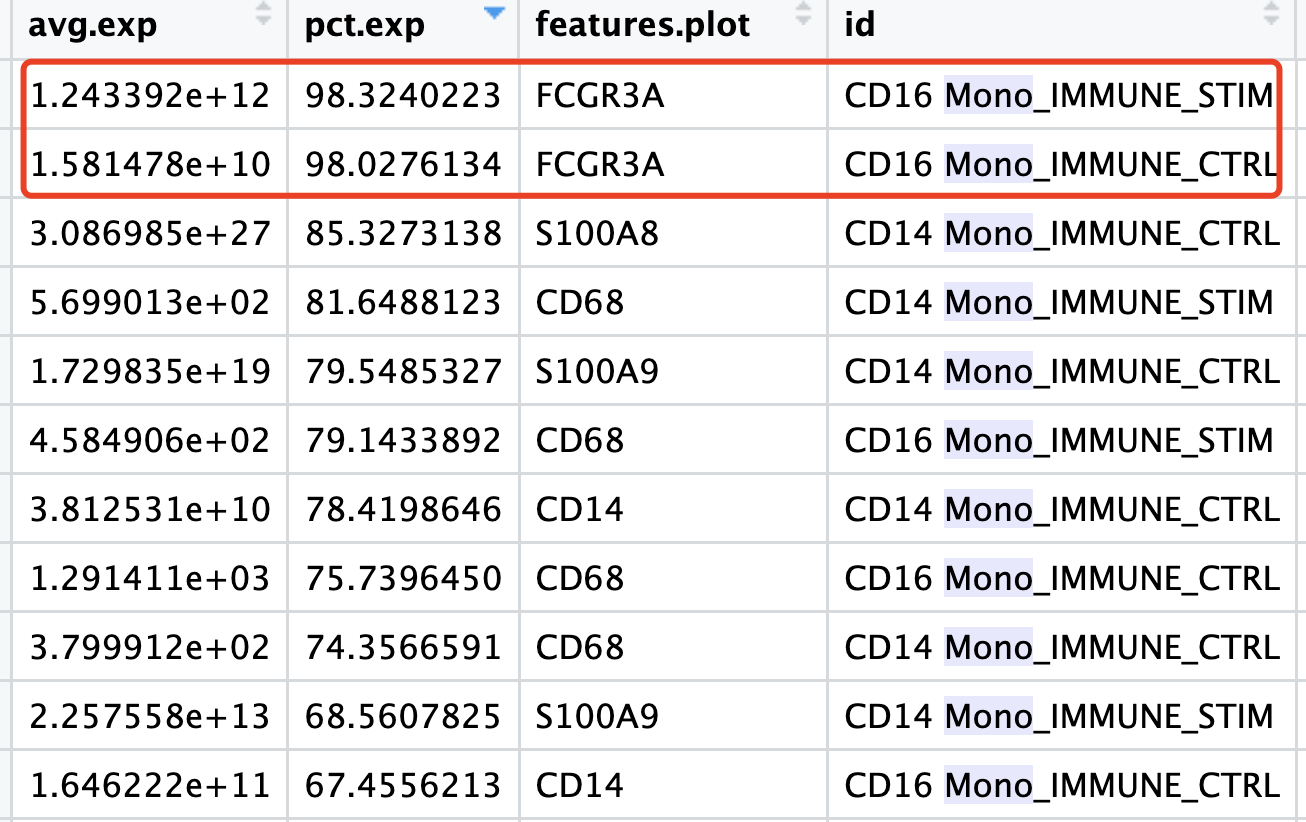
其实函数都是有参数的,上面的DotPlot函数就可以调整:
DotPlot(
object,
features,
assay = NULL,
cols = c("lightgrey", "blue"),
col.min = -2.5,
col.max = 2.5,
dot.min = 0,
dot.scale = 6,
留给大家做学徒作业吧,如何巧妙的使用DotPlot函数来可视化CD14和CD16的单核细胞的基因。
看到了现象还不够,还可以细究里面的原因
为什么在两个样品(两个处理)的单细胞转录组表达量矩阵里面,CD14和CD16的单核细胞的基因会总体来说在STIM组里面的远高于CTRL组呢?