单细胞转录组已经火爆了五个年头,到如今的2024大家其实已经非常熟悉了它的标准数据分析思路。都是先整合全部的样品的表达量矩阵后走降维聚类分群,然后第一层次降维聚类分群先在低分辨率的条件下对各个亚群进行生物学命名。如果是肿瘤领域的我们通常是进行如下所示的分类:
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibro or CD31+,PECAM1,endo)
参考我五年前介绍过的 CNS图表复现08—肿瘤单细胞数据第一次分群通用规则,这3大单细胞亚群构成了肿瘤免疫微环境的复杂。绝大部分文章都是抓住免疫细胞亚群进行细分,包括淋巴系(T,B,NK细胞)和髓系(单核,树突,巨噬,粒细胞)的两大类作为第二次细分亚群。但是也有不少文章是抓住stromal 里面的 fibro 和endo进行细分,并且编造生物学故事的。而且我们已经积累了心肝脾肺肾等多个器官的上皮细胞的细分亚群, 以及免疫细胞里面的髓系和B细胞细分亚群:
在针对各个细胞亚群进行细分的时候,大家也整理好了标准的分析条目,以2021的这个文章:《Single-cell RNA-Seq reveals transcriptional heterogeneity and immune subtypes associated with disease activity in human myasthenia gravis》为例,可以看到它里面的B细胞细分之后,可以进行更加细致的亚群命名,参考:B细胞细分亚群,然后每个亚群有自己的特异性基因,特异性的生物学功能 :
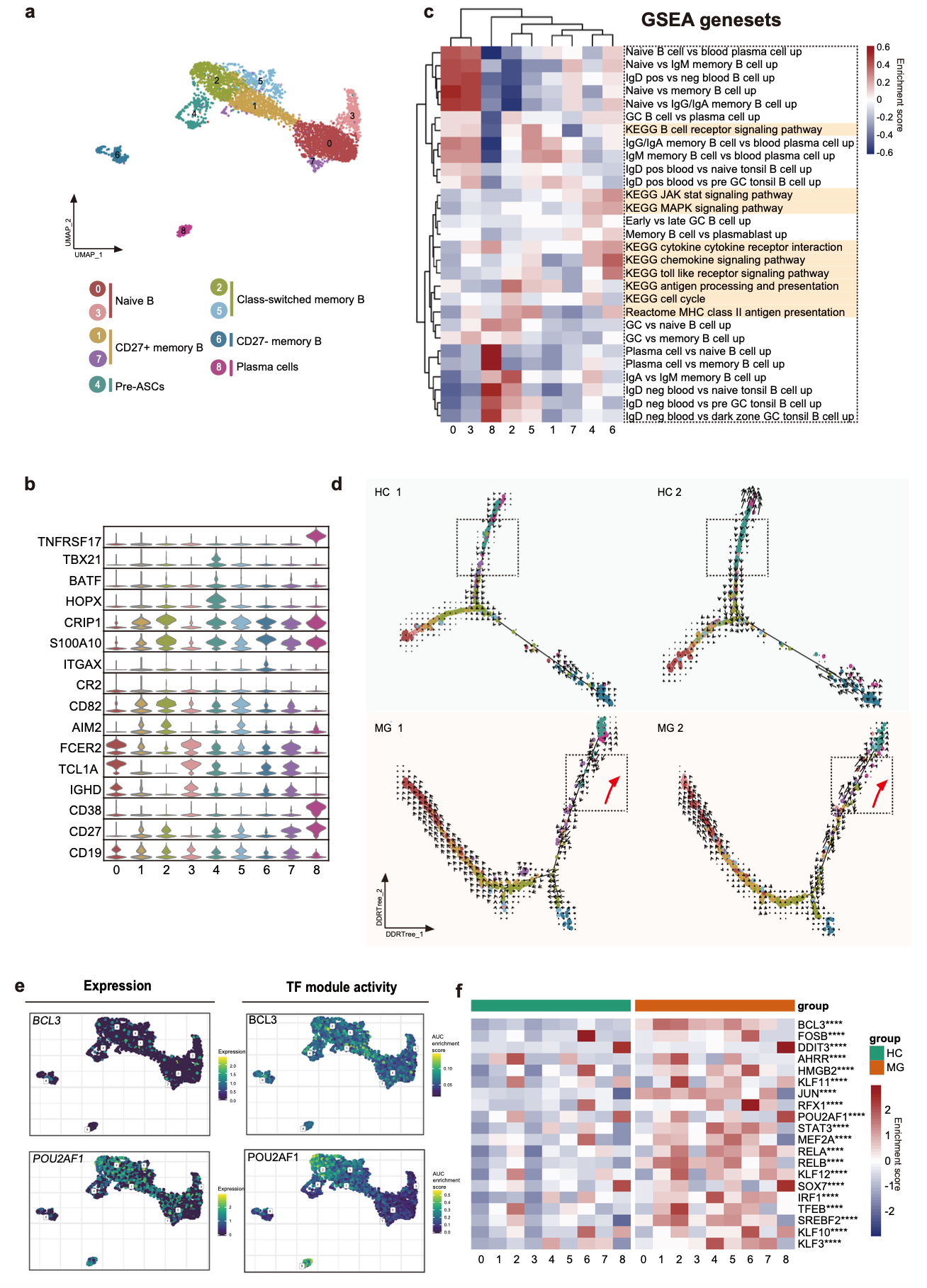
因为这些细分亚群都是b细胞,所以原则上靠通路富集有时候是不够描述各个亚群的特性的,尤其是在那些生物学背景模糊不清的时候。比如如果我们继续细分上面的b细胞里面的plasma细胞,这个时候如果算法继续给我们多个不同亚群我们就很难给出来生物学名字,而仅仅是按照编号来区分它们。我们仍然是针对不同编号的亚群进行算法层面的特异性基因或者通路进行功能学描述,但是略显不够。所以如下所示,我们增加了两个有挑战性的分析,就是拟时序和转录因子分析。
上面的案例,其实就回答了我们今天推文的问题:为什么做拟时序,因为它已经是非常常规而且几乎是必备的单细胞分析条目,你很难跳过去。而且,加入拟时序分析有时候确实是能更加说明问题。
有了差异分析还不够
既然我们可以针对均一性比较好的亚群细化后的不同编号的亚群进行算法层面的特异性基因或者通路,那么为什么我们还要做拟时序呢,其实特异性基因靠的是差异分析,是不同细胞亚群直接的差异分析,它这个差异分析的单位是细胞亚群而不是具体的每个细胞本身。实际上我们的单细胞转录组的优点应该是到单细胞分辨率,但是我们绝大部分的分析其实是仅仅是到了细胞亚群的分辨率,两个细胞亚群之间肯定是有各自的差异基因列表,但是因为这些细胞亚群是来自于一个均一性比较好的亚群所以 它们其实细化的时候很难有绝对的分界线,或者说它们之间就算是有分界线这个分界线也是可以打破的。
让我们看看具体的案例,众所周知CD14和CD16是用于表征单核细胞(单核吞噬细胞)亚型的两种常见标记物,CD14单核细胞主要参与炎症调节和免疫应答,而CD16单核细胞主要参与细胞毒性和抗体介导的免疫应答。而CD56和CD16这两种常用的表面标记物,用于区分NK细胞的亚型。我们可以很容易通过差异分析取找到CD14和CD16的单核细胞的各自的特异性基因列表,或者CD56和CD16的NK细胞的各自的特异性基因列表。如下所示是一个案例,我们这里是直接使用最简单的示范数据,来自于SeuratData包的ifnb数据集 :
library(Seurat)
library(SeuratData)
# InstallData('ifnb.SeuratData')
# 使用上面的代码下载SeuratData包的ifnb数据集,但是非常考验网络。。。。
# trying URL 'http://seurat.nygenome.org/src/contrib/ifnb.SeuratData_3.1.0.tar.gz'
# Content type 'application/octet-stream' length 413266233 bytes (394.1 MB)
# 如果网络不行,就想办法下载(394.1 MB)保存在自己的电脑里面,然后下面的代码安装指定路径的压缩包:
# install.packages(pkgs = '~/database/seurat-data/ifnb.SeuratData_3.1.0.tar.gz',
# repos = NULL,
# type = "source" )
# 一定要看到这个包成功安装哦
# * DONE (ifnb.SeuratData)
library(ifnb.SeuratData)
data("ifnb")
ifnb
ifnb=UpdateSeuratObject(ifnb)
ifnb
table(ifnb$orig.ident)
sce.all <- ifnb
有了上面的练习的数据集,接下来可以根据数据集自带的细胞亚群注释,从里面去挑选我们想做拟时序分析的单核细胞 (包括CD14和CD16的单核细胞);
table(sce.all$seurat_annotations)
sce.all$celltype = sce.all$seurat_annotations
sel.clust = "celltype"
scRNA <- SetIdent(sce.all, value = sel.clust)
table(scRNA@active.ident)
# 节省计算机资源
scRNA = subset(scRNA,downsample=100)
kp = scRNA$celltype %in% c('CD14 Mono' , 'CD16 Mono')
scRNA=scRNA[,kp]
table(scRNA$celltype)
对于CD14和CD16的单核细胞两个细胞亚群上面的组成的对象,我们很容易做差异分析
library(tidyverse)
markers <- FindAllMarkers(scRNA, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
all.markers =markers %>% dplyr::select(gene, everything()) %>% subset(p_val<0.05)
dim(all.markers)
table(all.markers$cluster)
top10 =all.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_log2FC)
tmp=ScaleData(scRNA,top10$gene)
DoHeatmap(tmp,features = top10$gene,raster = F)
可以看到的是CD14和CD16的单核细胞这两个细胞亚群确实是可以比较好的区分,但是它们也有融合的趋势,或者说有渐变的可能性,这个就是差异分析的细节问题。
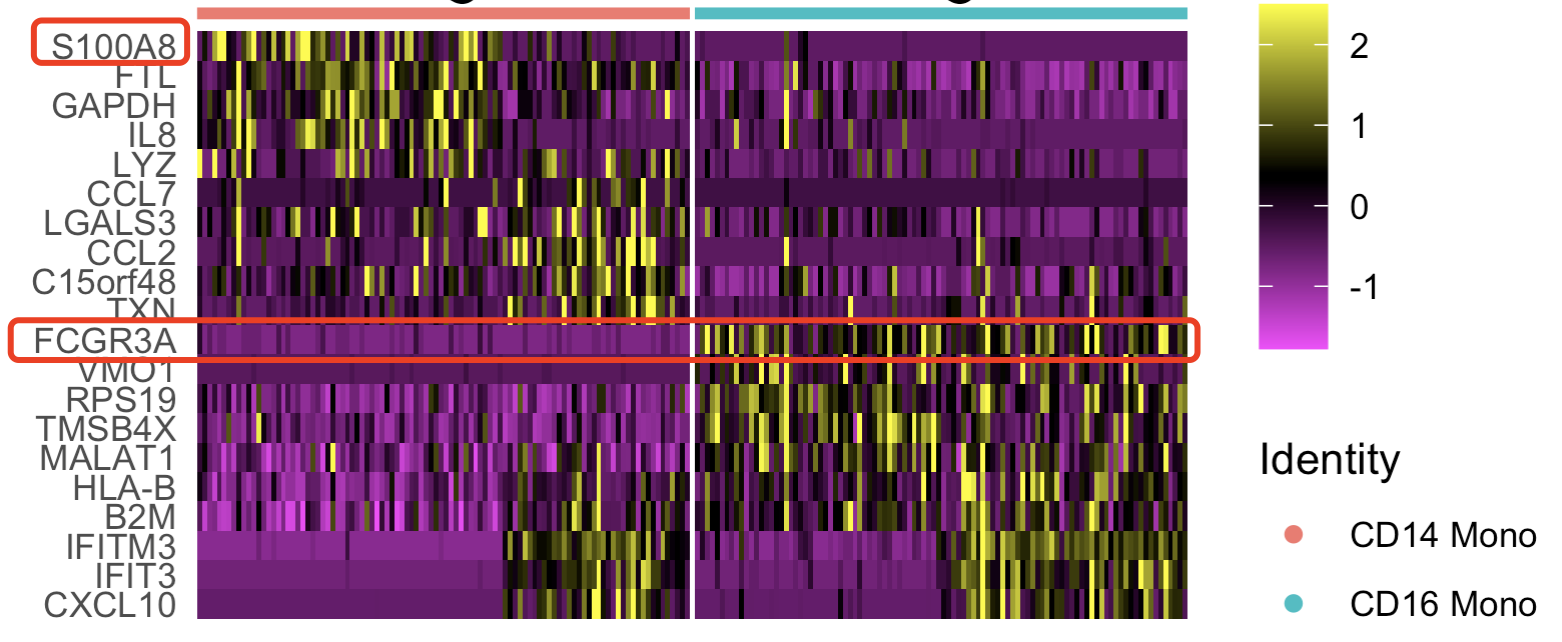
简简单单的差异分析当然是计算出来具体的基因在不同细胞亚群的变化倍数,统计学指标即可。
而要展示差异分析的细节,就不能是以细胞亚群为单位,而需要具体到每个细胞本身,去看看具体的基因是如何在这两个单细胞亚群的具体的细胞之间慢慢的变化。
尝试一下monocle2的拟时序分析
拟时序有非常多多种算法,后面我们再慢慢介绍,包括但不限于:
- monocle2拟时序实战
- monocle3拟时序实战
- SCORPIUS
- Slingshot
- 基于其它编程语言的拟时序分析
现在基于前面的CD14和CD16的单核细胞两个细胞亚群上面的组成的对象,我们尝试一下monocle2的拟时序分析,首先是构建monocle2的对象;
seurat=scRNA
library(monocle)
expr_matrix=seurat@assays$RNA$counts#使用counts表达值
sample_sheet<-seurat@meta.data#将实验信息赋值新变量
gene_annotation=data.frame(gene_short_name=rownames(seurat))#构建一个含有基因名字的数据框
rownames(gene_annotation)=rownames(seurat)#将上述数据框的行名赋值基因名字
pd <- new("AnnotatedDataFrame", data = sample_sheet)#将实验信息变量转化为monocel可以接收的对象
fd <- new("AnnotatedDataFrame", data = gene_annotation)#将基因注释变量转化为monocle可以接收的对象
cds <- newCellDataSet(expr_matrix, phenoData = pd, featureData = fd,
expressionFamily=negbinomial.size())#创建一个monocle的对象
cds #cellData对象;monocle独有
cds <- estimateSizeFactors(cds)
cds <- estimateDispersions(cds)
有了上面的monocle2的对象,拟时序只需要3个步骤;
# 第一步: 挑选合适的基因. 有多个方法,
# 例如提供已知的基因集,或者自己做差异分析后拿到的统计学显著的差异基因列表
diff_test_res <- differentialGeneTest(cds,
fullModelFormulaStr = " ~ celltype + orig.ident",
reducedModelFormulaStr = " ~ orig.ident",
relative_expr=TRUE, cores=4)
ordering_genes <- row.names (subset(diff_test_res, qval < 0.01))
ordering_genes
cds <- setOrderingFilter(cds, ordering_genes)
plot_ordering_genes(cds)
# 第二步: 降维。降维的目的是为了更好的展示数据。函数里提供了很多种方法,
# 不同方法的最后展示的图都不太一样, 其中“DDRTree”是Monocle2使用的默认方法
cds <- reduceDimension(cds,
max_components = 2,
num_dim = 6,
reduction_method = 'DDRTree',
residualModelFormulaStr = "~orig.ident", #去除样本影响
verbose = F)
# 第三步: 对细胞进行排序
cds <- orderCells(cds)
# 最后进行可视化
plot_cell_trajectory(cds, color_by = "orig.ident") +
plot_cell_trajectory(cds, color_by = "celltype")
就可以看到,前面的CD14和CD16的单核细胞两个细胞亚群其实是来源于两个不同样品或者说两个分组,sle病人的处理组和对照组。而前面的普通差异分析并没有考虑这个样品效应问题,但是我们的拟时序考虑到了,如下所示:
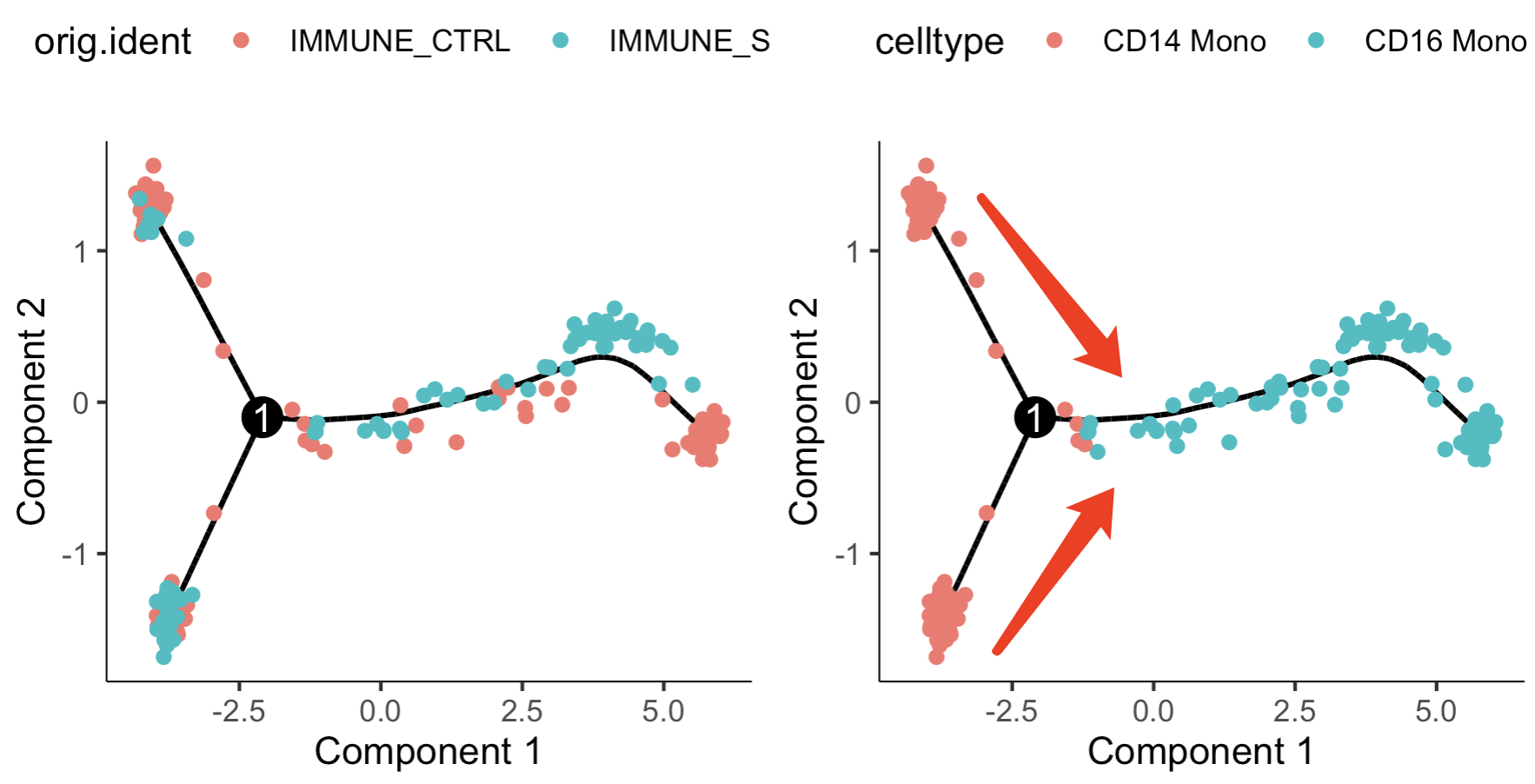
我们可以很清晰的看到,CD14的单核细胞其实是有两个截然不同的再细分的亚群,需要补充背景知识啦:
- https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2019.01761/full
- https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2020.01070/full
那么拟时序到底是得到了什么样的结果呢,我们可以继续可视化另外两个关键的变量,就是拟时序分析后产生的 Pseudotime 和State :
library(ggsci)
p1=plot_cell_trajectory(cds, color_by = "celltype") + scale_color_nejm()
p1
p2=plot_cell_trajectory(cds, color_by = "Pseudotime")
p2
p3=plot_cell_trajectory(cds, color_by = "State") + scale_color_npg()
p3
library(patchwork)
p1+p2/p3
可以看到,如果是拟时序的算法层面来说,是CD16的单核细胞慢慢演变成为了两种截然不同的CD14的单核细胞,如下所示:
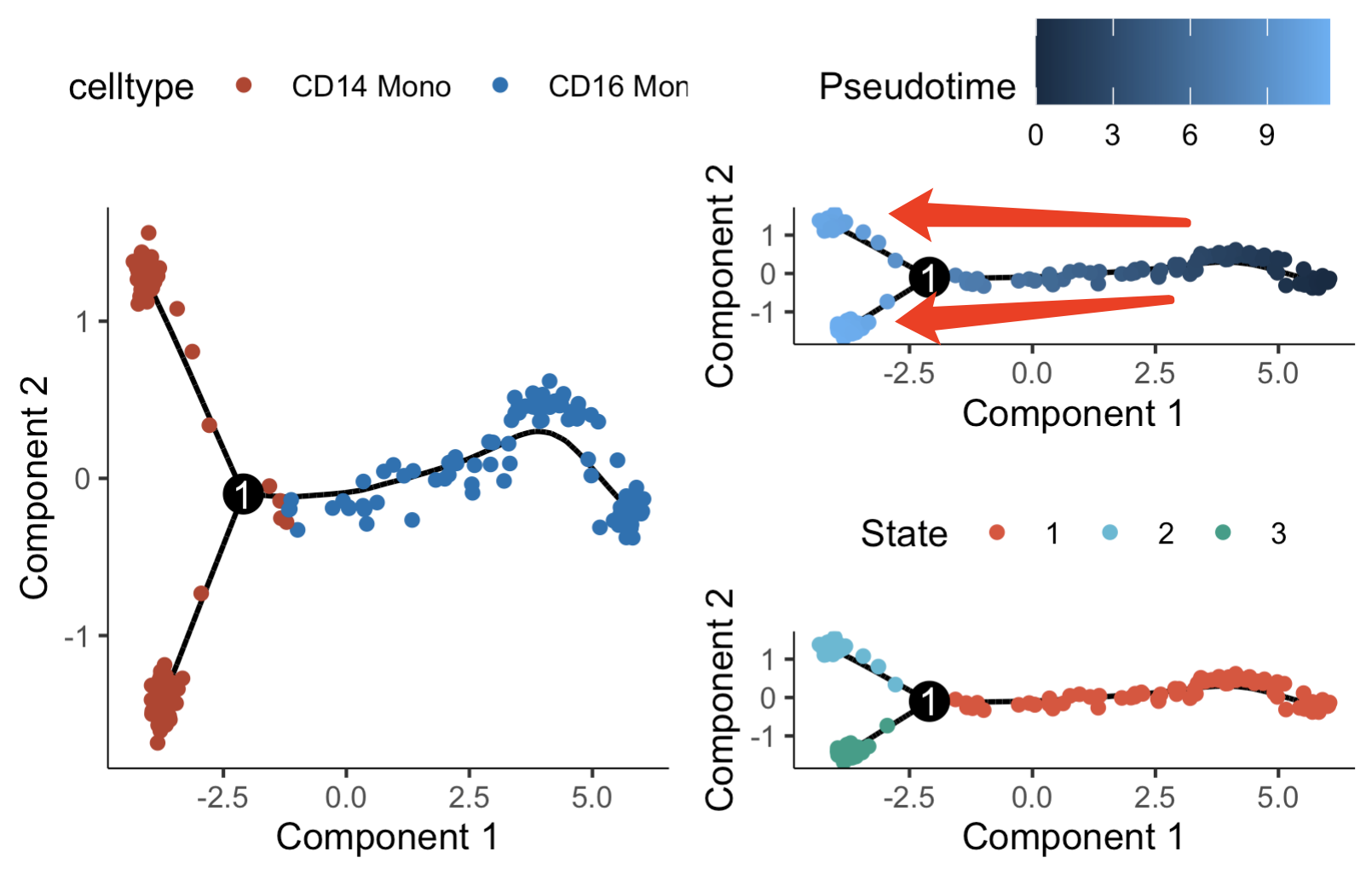
值得注意的是,这个演变的顺序其实可以认为颠倒过来的,因为算法推断其实没办法知道发育顺序,仅仅是有有一些基因验证拟时序在变化。我们可以有很多进一步的分析的可能性,就需要熟练的掌握拟时序分析后针对每个细胞 产生的 Pseudotime数值和State分组 ,如下所示 :
> colnames(pData(cds))
[1] "orig.ident" "nCount_RNA" "nFeature_RNA" "stim"
[5] "seurat_annotations" "celltype" "Size_Factor" "Pseudotime"
[9] "State"
> head(pData(cds))
orig.ident nCount_RNA nFeature_RNA stim seurat_annotations celltype
AAAGGCCTAAACGA.1 IMMUNE_CTRL 2675 792 CTRL CD16 Mono CD16 Mono
AAATACTGGTTCTT.1 IMMUNE_CTRL 2922 830 CTRL CD14 Mono CD14 Mono
AAATCCCTGTTGAC.1 IMMUNE_CTRL 2139 640 CTRL CD14 Mono CD14 Mono
AAATTGACAAACAG.1 IMMUNE_CTRL 1854 716 CTRL CD14 Mono CD14 Mono
AACTCTTGCATTCT.1 IMMUNE_CTRL 2219 764 CTRL CD16 Mono CD16 Mono
AAGACAGAGTTAGC.1 IMMUNE_CTRL 1760 619 CTRL CD14 Mono CD14 Mono
Size_Factor Pseudotime State
AAAGGCCTAAACGA.1 1.0390142 5.7395392 1
AAATACTGGTTCTT.1 1.1349531 10.9696635 3
AAATCCCTGTTGAC.1 0.8308230 7.6699632 1
AAATTGACAAACAG.1 0.7201243 10.8909299 3
AACTCTTGCATTCT.1 0.8618963 0.1031971 1
AAGACAGAGTTAGC.1 0.6836131 10.5724209 2
> table(pData(cds)$State)
1 2 3
105 49 46
而且也可以把这个拟时序分析后针对每个细胞 产生的 Pseudotime数值和State分组 对象回帖到常规的Seurat的降维聚类分群流程里面去可视化,简单的代码如下所示:
sce <- SCTransform(scRNA,verbose = F)
sce <- RunPCA(sce,verbose = F)
sce <- RunUMAP(sce, dims = 1:20)
DimPlot(sce,group.by = 'celltype')
sce$State = pData(cds)$State
sce$Pseudotime = pData(cds)$Pseudotime
DimPlot(sce,group.by = 'State') +
DimPlot(sce,group.by = 'celltype') +
DimPlot(sce,group.by = 'orig.ident')
可以看到,在二维平面上面,CD14和CD16的单核细胞两个细胞亚群的差异其实是跟两个不同样品或者说两个分组并存的,而且是在每个样品内部都很清晰的可以看到是CD16的单核细胞慢慢演变成为了两种截然不同的CD14的单核细胞。
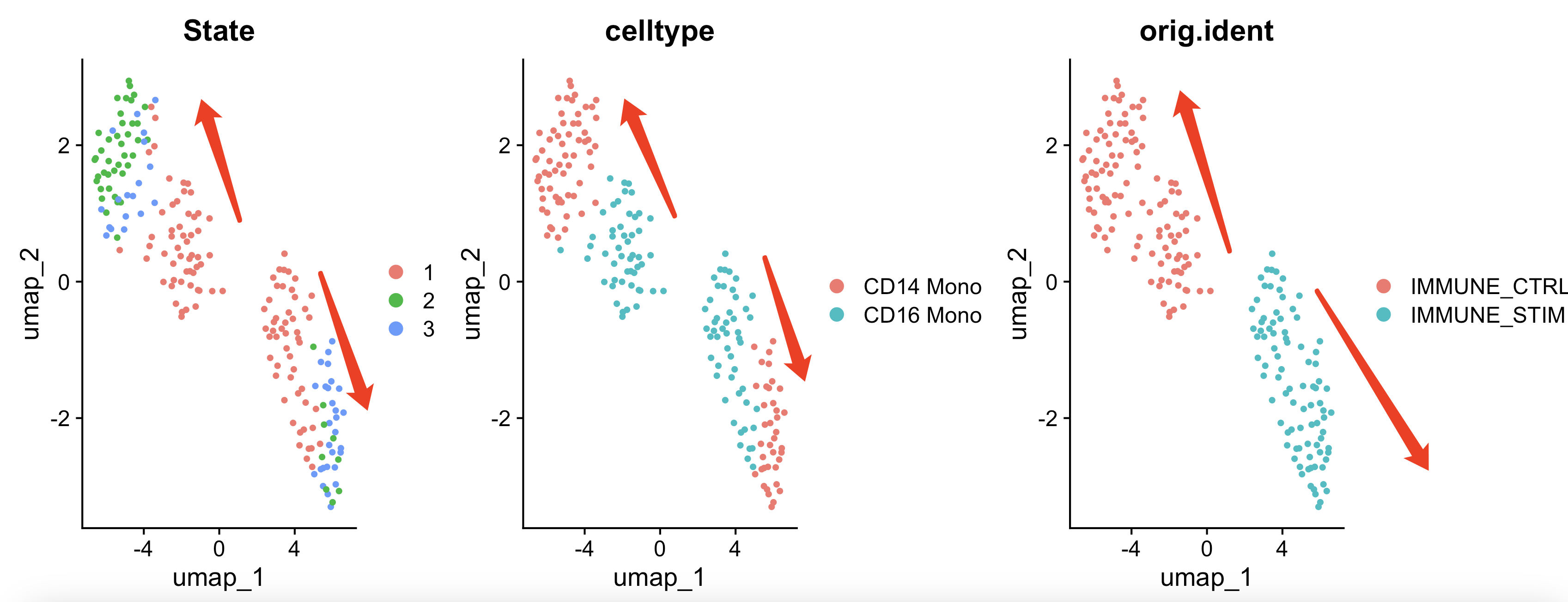
这个具体的变化过程,就可以继续两两之间做拟时序看基因列表了,绘制渐变的热图。
拟时序其实也不够
虽然上面的案例说明了拟时序分析是常规差异分析的很好的补充,把分辨率从细胞亚群为单位提高到了具体的单个细胞本身,因为每个细胞都有自己的唯一的 Pseudotime数值,根据这个数值就可以对细胞进行排序啦。但是这个算法本身也是不够的,它得到的 Pseudotime数值是大小的排序是可以反过来的,这个时候需要一些先验知识的辅助,比如某些基因是已知的的顺序变化趋势。又或者需要一些其它算法的辅助,比如 使用cytoTRACE评估不同单细胞亚群的分化潜能。
当然了,如果超脱表达量矩阵本身,引入全新信息,比如具体的基因的转录本的未剪接和剪接 mRNA数值,就可以构建RNA 速率模型做分析,得到的发育顺序就有方向而且可靠啦,详见:10x官网下载pbmc3k数据集走RNA速率上下游分析实战。但是它也有缺点,首先是需要对测序的fastq文件重新分析,才能获取到未剪接和剪接 mRNA数值,其次在传统的基于二代测序数据的10x单细胞转录组其实仅仅是测了具体的每个mRNA的两端二选一而已,并没有把两个mRNA测通所以就很难获得准确的未剪接和剪接 mRNA数值。
当然了,算法会一直有更新,全新的软件和工具也会被持续不断的开发出来,慢慢的就解决了以前的问题。比如2024新发表的scVelo,就不再依赖于剪接信息,而是通过引入基因调控数据 ,提供更准确的细胞轨迹信息和伪时间推断结果。详见
- 文章:TFvelo: gene regulation inspired RNA velocity estimation. Nat Commun
- github官网(Python软件):https://github.com/xiaoyeye/TFvelo
你现在看到的是拟时序一本通专辑
今天我们分享了为什么做拟时序,把它简化成为了主要是为了展示差异细节这个功能,忽略了它在发育学层面的应用,因为我不是发育生物学领域背景。虽然说拟时序分析在单细胞转录组数据分析中具有重要意义,可以帮助我们理解细胞状态的动态变化和发展轨迹,以及疾病的发展过程和治疗机制,主要是有下面的这些应用,需要结合具体的文献来解读:
- 识别细胞状态的动态变化: 单细胞转录组数据通常是通过捕获大量单个细胞的基因表达数据来获取样品中细胞的多样性。拟时序分析可以帮助我们理解细胞在不同时间点或条件下的动态变化,识别细胞状态的转变过程,比如细胞分化、激活、增殖等。
- 恢复发育轨迹: 拟时序分析可以帮助恢复细胞发育轨迹,揭示细胞从干细胞到成熟细胞的分化过程。通过构建细胞的发育轨迹图,可以将细胞按照它们在发育过程中的相似性进行聚类,进而研究发育过程中的关键转变和调控因子。
- 研究疾病进展过程: 在疾病研究中,拟时序分析可以帮助我们理解疾病的发展过程,识别疾病相关的细胞亚型或状态的动态变化,以及发病机制的关键节点。这对于疾病诊断、预测和治疗具有重要意义。
- 探索细胞在特定条件下的响应机制: 拟时序分析可以帮助我们研究细胞在不同环境或刺激条件下的响应机制。通过比较不同时间点或条件下的细胞表达模式,可以识别出响应于特定刺激的基因表达变化,并揭示其潜在的调控网络和信号通路。
值得一提的是拟时序一本通专辑的目录是:
- 为什么做拟时序(展示差异细节)
- 拟时序的正确姿势,错误示范,创新型拟时序
- 拟时序的多种算法大比拼
- monocle2拟时序实战
- 降维聚类分群
- 拟时序
- 多种基础可视化
- 正向特殊可视化(目标基因表达量,基因集打分)
- 反方向特殊可视化(5种可视化)
- monocle3拟时序实战
- SCORPIUS实战
- Slingshot实战
- 基于其它编程语言的拟时序分析
然而,本人的理解肯定是片面的,希望通过这10个推文的指引能让大家对单细胞转录组的明星分析方法拟时序有一个基础的认知。也欢迎大家提出来自己的不同的看法,或者自己对拟时序的疑惑之处,欢迎留言参与讨论哦!